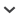Gene flow in plants is mediated by processes that cause changes in allele frequencies, including movement of gametes (gametophytes) or individuals (sporophytes) across the physical landscape (Slatkin, 1987). Movement by gametes occurs by dispersal of pollen (the male gametophyte) from the location of the pollen donor to the pollen recipient. In the sporophytic life stages, plants are either sessile or have limited mobility through vegetative growth, and dispersal of individuals is reduced to movement of vegetative propagules or seeds. Seed, propagule, and pollen dispersal contribute to gene flow, but of these, only dispersal of sporophytes has the potential to establish new populations through colonization of vacant sites (Howe and Smallwood, 1982; Nathan and Muller-Landau, 2000).
Both pollen and seed dispersal contribute to gene flow and, consequently, affect the distribution of genetic variation within and among populations; however, population genetic studies rarely consider the separate effects of the movement of sporophytes and gametophytes on population genetic structure (e.g., Ennos, 1994; McCauley, 1994; Ouborg et al., 1999). Seed dispersal is not only important for gene flow, but also responsible for the colonization of new sites and range expansion. Ecological approaches to the measurement of seed dispersal can be difficult to implement and tend to overestimate short-distance seed dispersal while failing to detect long-distance dispersal events (Willson, 1993). Long-distance seed dispersal may be disproportionately important for gene flow and establishing new populations (Cain et al., 2000; Trakhtenbrot et al., 2005). Maternally inherited genetic markers can be used to measure the influence of historical dispersal on gene flow and to make inferences about population history through the application of phylogeographic analyses (Cruzan and Templeton, 2000; Knowles, 2009; Nielsen and Beaumont, 2009). Chloroplast DNA (cpDNA) is inherited maternally in most angiosperms (Corriveau and Coleman, 1988), which means variation in these markers is only affected by the process of seed dispersal.
In the past, cpDNA markers have not been considered very useful due to the slow evolutionary rate of chloroplast genomes, which results in low intraspecific variation (Palmer, 1987). This was particularly true for traditional methods for assaying sequence variation using restriction enzymes (e.g., McCauley, 1994; Soltis et al., 1997; Maskas and Cruzan, 2000). Modern sequencing methods combined with targeted capture alleviate this problem by allowing the detection of larger numbers of sequence variants (single-nucleotide polymorphisms [SNPs]) across the entire chloroplast genome (Stull et al., 2013). Combinations of SNP alleles represent chloroplast haplotypes and are a valuable tool for examining genetic diversity and inferring historical patterns of dispersal and migration.
Using cpDNA variation (in the form of cpDNA SNPs) to measure genetic levels of genetic differentiation presents a few challenges. First, chloroplast genomes are nonrecombining and effectively haploid (Palmer, 1987), so SNP alleles common to the same haplotype are inherited together. This allows for the reconstruction of network phylogenies that illustrate the relationships among haplotypes, but it also means that, no matter how many cpDNA SNPs are found, the whole chloroplast can only be treated as a single locus. We found that treating cpDNA SNPs as independent markers tends to underestimate levels of differentiation and genetic distances among populations, especially when haplotypes share many SNP alleles (Fig. 1).
When using chloroplast haplotypes for population genetic and phylogeographic studies, cpDNA from many individuals must be sequenced to generate adequate sample sizes for the estimation of genetic parameters. Although sequencing costs have decreased in recent years, sequencing enough samples for large-scale population genetic and phylogeographic studies still requires a significant resource investment (Sboner et al., 2011). Pooling multiple individuals for sequencing has become a common solution to this problem (Sham et al., 2002; Schlötterer et al., 2014). Unfortunately, pooling cpDNA samples results in the loss of information about the SNP allele associations that represent each haplotype because DNA sequencing only recovers SNP allele frequencies (Fig. 1). Although there are a number of haplotype reconstruction programs available, these are either aimed exclusively at diploid genomes or at resolving (nuclear) haplotypes over smaller genomic regions (i.e., phasing; Pe'er and Beckmann, 2003; Kirkpatrick et al., 2007; Gasbarra et al., 2011; Kofler et al., 2011). These methods assume some level of recombination and, ultimately, are not appropriate for the recovery of haplotypes from the nonrecombining chloroplast genome. To solve this problem, we have developed a new sample preparation and bioinformatics pipeline (Fig. 2) aimed at reducing the cost of population-level surveys of chloroplast diversity by reconstructing chloroplast haplotypes from pooled samples from an initial sample of sequenced individual chloroplast haplotypes.
Fig. 1.
SNP frequency contribution from multiple haplotypes where a SNP is shared between haplotypes. In this case, each population contains the same three haplotypes, with one being found at a constant frequency in all three populations, while the other two, which share a SNP allele, are found at varying frequencies in the three populations, such that the overall frequency of that SNP is constant. A network phylogeny showing the three haplotypes and their relatedness to each other is shown below the figure. Haps = haplotype.

Here, we describe sampling and bioinformatics protocols for the examination of haplotype-based population genomics and phylogeography (CallHap), which includes programs that conduct variant filtering, haplotype recovery, assembly of network phylogenies, and the estimation of haplotype frequencies from pooled samples. We then test the CallHap haplotype recovery program using a series of artificial networks and pools. Finally, we provide an example of CallHap processing using a set of Lasthenia californica DC. ex Lindl. (Asteraceae) samples collected from Whetstone Savanna Preserve near Medford, Oregon, USA.
THE CALLHAP PIPELINE
Experimental design, sampling, and sequencing library preparation—Experiments designed to use the CallHap pipeline need to be planned carefully. First, multiple individuals from some number of populations or locations are sampled, and whole genomic DNA isolated and purified. Pools are constructed using equimolar amounts of DNA. We suggest a pool size of 20, but this can be adjusted based on the goals of the study (for more detail, see the discussion). Two types of sequencing libraries are prepared: (1) a representative sample of individual libraries (single-sample libraries [SSLs]) from across the range and (2) a set of pooled libraries (PLs). The SSL haplotypes are used to establish a skeleton phylogeny, so sampling should be adjusted to attempt to capture a wide range of variation. The PLs will be used to identify new haplotypes and to estimate pooled haplotype frequencies. As a general guideline, the number of SSLs should be about the same as the number of PLs. If there is reason to suspect large amounts of divergence between sampled populations, additional artificial pools containing individuals from different portions of the range should also be constructed (see explanation of artificial pools in the discussion).
Fig. 2.
Overview of sampling, labwork, and bioinformatics protocols involved in a CallHap experiment.

Libraries (both PLs and SSLs) are multiplexed for cpDNA targeted capture and sequencing. Equimolar contributions are used to combine up to 60 libraries for paired-end sequencing on a single lane of the Illumina 2500 HiSeq (Illumina, San Diego, California, USA) or equivalent instrument (multiplex size should be adjusted for instruments with different capacities). Chloroplast genomic DNA is captured from multiplexed libraries prior to sequencing using custom-designed RNA bait arrays (e.g., Stull et al., 2013; see below). Bioinformatics processing and filtering prior to CallHap analysis are described in Appendix S1 (apps.1700053_S1.pdf).
Variant filtering—Variant filtering is accomplished using the first of the two CallHap programs, CallHap_VCF_Filt.py (see the program flowchart in Appendix S2 (apps.1700053_S2.pdf)). This script filters raw variants. To ensure that they can be used by the main haplotype caller, the following variants are removed: (A) non-SNP variants (due to the difficulty in calling insertion- or deletion-type variants [indels] as being in one of two states), (B) variants with low depth or quality, (C) variants that do not have a defined identity across all SSLs and PLs (because the haplotype caller application cannot handle missing values in the matrix of haplotype identities), and (D) SNPs in close proximity to indels (due to difficulties in creating correct alignments in these regions). Filters have a limit (depth filter, indel proximity, and quality filters) that can be modified by the user to meet the demands of a particular study. The variant filter outputs a file containing genotype data at all SNP loci for SSLs (Haps file), a separate file containing SNP reference allele frequency data for PLs (Pools file), and a NEXUS file for network phylogeny creation.
Haplotype identity and frequency determination—The CallHap Haplotype Caller (CallHap_HapCallr.py, Appendix S3 (apps.1700053_S3.pdf)) works by iterating through all the available SNPs in a pseudorandom order, with polymorphic SNPs in SSL (known) haplotypes being processed first. Processing a large number of these random orders increases certainty in haplotype calls. Within each order, the CallHap Haplotype Caller uses a least squares algorithm ( Appendix S4 (apps.1700053_S4.pdf)) to solve the equation  for the minimum residual sum of squares (RSS) value, where A is the binary (1's and 0's) n* m matrix of haplotypes, bi is the n*1 vector of observed SNP frequencies in the ith pool,
for the minimum residual sum of squares (RSS) value, where A is the binary (1's and 0's) n* m matrix of haplotypes, bi is the n*1 vector of observed SNP frequencies in the ith pool,  (bi) is an estimator for bi, and xi is the 1 *m estimated vector of haplotype frequencies in the ith pool. Within the haplotypes matrix A, each column represents a haplotype and each row represents a SNP locus; the value in that particular element of the haplotypes matrix indicates which allele of the given SNP is present in the given haplotype. When solving this equation within a round of haplotype estimation (A remains constant), each xi is chosen such that
(bi) is an estimator for bi, and xi is the 1 *m estimated vector of haplotype frequencies in the ith pool. Within the haplotypes matrix A, each column represents a haplotype and each row represents a SNP locus; the value in that particular element of the haplotypes matrix indicates which allele of the given SNP is present in the given haplotype. When solving this equation within a round of haplotype estimation (A remains constant), each xi is chosen such that  is minimized. Initially, A is composed entirely of haplotypes observed in the SSLs (as defined in the Haps file), but in later rounds of haplotype estimation, A expands to contain estimated novel haplotypes in addition to the initial haplotypes. All instances of bi are read from the Pools file produced by the VCF filter.
is minimized. Initially, A is composed entirely of haplotypes observed in the SSLs (as defined in the Haps file), but in later rounds of haplotype estimation, A expands to contain estimated novel haplotypes in addition to the initial haplotypes. All instances of bi are read from the Pools file produced by the VCF filter.
In each round of haplotype estimation, several values of A (each containing a different estimated haplotype) are tested. When creating new haplotypes, a SNP is only considered if there exists a nonzero residual in the current solution for that SNP locus ( Appendix S5 (apps.1700053_S5.pdf)). If the current SNP is polymorphic in A, new haplotype creation only considers creating new haplotypes based on the haplotypes at either end of the network phylogeny branch along which this SNP occurs. Otherwise, the algorithm considers every possible new haplotype (Fig. 3). At the end of each round, only those values of A with the lowest  are kept for further rounds of haplotype estimation (Fig. 3). An example of the matrices is shown in Appendix S6 (apps.1700053_S6.pdf). Once all SNPs have been processed, the haplotypes matrices are filtered to remove unused haplotypes. Haplotypes matrices are then filtered to keep only those with the lowest Akaike information criterion (AIC; Li et al., 2002). The columns of these matrices (the haplotypes) are taken as binary numbers, with 1 representing the reference and 0 the alternate allele, converted into decimal numbers representing the haplotypes, and saved along with the average RSS values produced by the matrices.
are kept for further rounds of haplotype estimation (Fig. 3). An example of the matrices is shown in Appendix S6 (apps.1700053_S6.pdf). Once all SNPs have been processed, the haplotypes matrices are filtered to remove unused haplotypes. Haplotypes matrices are then filtered to keep only those with the lowest Akaike information criterion (AIC; Li et al., 2002). The columns of these matrices (the haplotypes) are taken as binary numbers, with 1 representing the reference and 0 the alternate allele, converted into decimal numbers representing the haplotypes, and saved along with the average RSS values produced by the matrices.
After completing all pseudo-random orders, output files are generated showing the raw haplotypes produced in each proposed solution, the percentage of random orderings for which a particular haplotype was produced, the number of times each unique topology was generated, and the average RSS value for each. In addition, the following files are generated: files containing haplotype frequencies in each pool and the RSS value for that pool, VCF files showing predicted SNP reference allele frequencies in each pool and RSS for each SNP, a CSV file comparing observed vs. predicted SNP frequencies, and a NEXUS file for examining the proposed network phylogeny. Optionally, a genpop file that can be imported into adegenet (Jombart, 2008) and a STRUCTURE-formatted file (Pritchard et al., 2000; Raj et al., 2014) can be generated. Haplotype frequencies are represented as number of individuals in the pool with that haplotype, and haplotypes are represented as multiple alleles at a single locus (the chloroplast).
Fig. 3.
Haplotype creation and selection of best position in a simple haplotype system. In each case, N represents the position of the newly created haplotype. Graphs show predicted vs. observed reference allele frequencies for SNPs. RSS = residual sum of squares.

After CallHap generates outputs, users can examine the resulting topologies and select a final topology based on (1) the average RSS value of the solution, (2) the frequency with which a given topology occurred, and (3) the commonality of the root haplotype for any ambiguous new haplotypes not resolved by the first two criteria (Templeton et al., 1992).
MATERIALS AND METHODS
Testing with artificial networks and pools—We tested the CallHap pipeline using a set of artificially created network phylogenies and pool frequencies. Test networks were created to represent different types of network topologies (Fig. 4). Seven artificial pools containing 20 individuals each were created based on each network, with each pool containing three random haplotypes at frequencies approximating the Poisson distribution. The Poisson distribution was chosen because it often reflects natural frequency distributions, and the results obtained were not sensitive to haplotype frequencies within pools. Each set of artificial pools was processed with the haplotype caller using 100 random orders, with two iterations per order and different combinations of “known” haplotypes to determine whether both the correct haplotype network phylogeny and haplotype frequencies were recovered by the best solution.
Testing with pooled population samples—Leaf tissue was collected from 400 individuals across 20 populations of L. californica located within a 16-ha area of Whetstone Savanna Preserve, near Medford, Oregon, USA (P. Thompson et al., unpublished data). Leaf tissue was dried using silica beads as a desiccant, and DNA was extracted using a QIAGEN Plant DNeasy 96 kit (QIAGEN, Germantown, Maryland, USA). After DNA extraction, DNA concentration was quantified on a Qubit 3.0 fluorometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and pooled by population in an equimolar fashion (20 samples per PL). Library preparation was conducted using a NEBNext Ultra DNA Library Prep Kit (E7370) with NEBNext Multiplex Oligos (E7600; New England Biolabs, Ipswich, Massachusetts, USA). SSLs were constructed for at least one individual from each population.
Fig. 4.
Test network phylogenies. These phylogenies were designed to test the ability of CallHap to recover different topological patterns when starting with different haplotypes: (A) a long branch with every haplotype defined, (B) two long branches with all haplotypes defined, (C) a long branch with some haplotypes defined, and (D) a cluster with one haplotype further out.

SSLs and PLs were captured using a MYbaits-3 custom cpDNA capture array from MYcroarray (Ann Arbor, Michigan, USA; Appendix S7 (apps.1700053_S7.docx)). DNA was sequenced on an Illumina HiSeq 2500 Sequencer (Illumina), with 100-bp paired-end reads generated for all but six samples, which had 100-bp single-end reads (Massively Parallel Sequencing Shared Resource Facility, Oregon Health and Science University, Portland, Oregon, USA). The contents of each lane are summarized in Table 1. Sequence alignment was performed to an inhouse L. californica chloroplast genome assembly (GenBank KY965816). SNP calling, variant filtering, and haplotype calling were performed using the pipeline as described above with a minimum read depth of 600 and a minimum variant quality of 20. Haplotype calling was performed using information from the L. californica sequence alignments. For the full data set, haplotype calling was run a second time with any new haplotypes that were consistently added placed in the input haplotypes to help resolve ambiguous haplotypes.
RESULTS
Test networks—Correct haplotype networks were recovered as single lowest RSS value solutions in all starting conditions for three out of four test networks. For the fourth, the correct haplotype network was recovered as the more common of two possible solutions with the lowest RSS value (Fig. 5).
Sequencing and variant calling—Sequencing performed for L. californica produced 67 libraries (47 SSLs and 20 PLs), which amounted to 753,355,673 raw reads. After variant calling, 978 initial variants were recovered, which simplified to 39 SNPs in 19 unique haplotypes after filtering. Initial haplotype calling produced two solutions at a minimum RSS value of 0.003002, with seven new haplotypes common to all the top three solutions and three ambiguous haplotypes. Rerunning CallHap with the common haplotypes added to the SSL haplotypes returned three solutions: one with an RSS value of 0.003002, one with an RSS value of 0.003077, and one with an RSS value of 0.003165 (these topologies are summarized in Fig. 6, and RSS values are summarized in Tables 2 and 3). Although the best RSS value solution was not the most common solution, the difference in the RSS values was small enough that the solutions are essentially equivalent. Additionally, there were only minor differences in haplotype frequency between the best RSS value solution and the second best RSS value solution. Because the RSS values for the best two solutions were effectively the same (i.e., within 5% of each other), the more common topology was selected as the best solution.
Table 1.
Summary of sequencing lane contents, showing number of Lasthenia californica single-sample libraries and pooled libraries used in analysis on each lane, number of other libraries on each lane, percentage L. californica returns from each lane, and type (single end or paired end) of each run.

DISCUSSION
We have developed a pipeline, CallHap, for efficient examination of cpDNA variation and tested it using a variety of test networks and a real data set of L. californica samples from Whetstone Savanna Preserve. Here, we present: (1) an examination of test results, (2) considerations for the design of experiments using CallHap, and (3) appropriate protocols for analysis of CallHap outputs. In addition, we provide an explanation for the magnitude of RSS values calculated by CallHap and a discussion of potential applications for this protocol.
Fig. 5.
Resulting phylogeny from one starting condition from Test Network D. (A) Green haplotypes were known at the beginning, blue haplotypes were present in all solutions at the lowest RSS value, and orange haplotypes had ambiguous positions between different solutions. Branch thickness is scaled by how many times a solution with the branch occurred, and percentages give exact percent of time a branch occurred. Hash marks indicate number of SNPs along a branch. (B) Regression plot for these solutions.

Test results—Examining the test network pools reveals consistent recovery of haplotype networks from a starting point of two or more haplotypes (SSLs) in the absence of any sequencing error. The presence of two possible solutions in the fourth test network reveals one potential problem that could arise during haplotype construction; if the frequency of a new haplotype is less than the frequencies of multiple other haplotypes across all PLs, the new haplotype may be placed ambiguously among multiple locations on the network. When the false haplotype position was not one of the known haplotypes, the correct solution was always the most common solution. One remedy to this issue would be to add new haplotypes that were found consistently among the solutions with the best RSS values to the starting haplotypes array, and then rerun the program as was done above with the Lasthenia data. By using the expanded array of haplotypes as a starting point, differences among solutions with the same RSS value may be resolved. Another method involves taking the source DNA samples and creating extra PLs by reshuffling the samples in ways that do not reflect the geographic areas in which the samples were collected (i.e., artificial pools [discussed in more detail later]).
Testing also revealed that, with minimal sampling of SSLs, convergence to a best solution was proportional to the centrality of the starting haplotype. As an example, for one of the test pools, all 100 orders converged to the lowest RSS value when the starting haplotype was the most central haplotype, as opposed to 13/100 and 3/100 for starting haplotypes one and two SNPs different from the most central haplotype, respectively. Furthermore, the presence of long branches in the correct topology reduced the frequency with which that topology came up. In cases where CallHap is finding a large number of topologies, it would be advisable to rerun CallHap with a larger number of random orderings along with augmenting the known haplotypes with any new haplotypes found universally. In addition, starting with more than one SSL per population (pool) sampled will increase the likelihood that the most central haplotype will be included in the SSL haplotypes.
It is apparent from examining the inferred haplotype frequencies for L. californica that RSS values for individual populations differ substantially. There can be many reasons for this. In some cases, high RSS values may be due to low-quality SNPs that escaped filtering. For this reason, even after automated SNP filtering, any remaining SNPs should be visualized using Integrative Genomics Viewer (IGV; Thorvaldsdóttir et al., 2013) or other similar programs to verify quality. Potential issues include SNPs that occur at approximately the same frequency across populations while the other SNPs in the pool change frequencies (especially if the major SNP present in the pool changes frequency). In these cases, the SNPs displaying consistent frequencies are most likely artificial and should be removed.
Another potential source of error is heteroplasmy (multiple chloroplast haplotypes within a single individual), which is caused by biparental inheritance of chloroplast genomes and somatic mutation. Past studies on heteroplasmy suggest that paternal inheritance occurs at a rate of about 1–2% (Cruzan et al., 1993; Ellis et al., 2008). Inferred haplotype frequencies for L. californica had very small errors around expected values (see below), suggesting that heteroplasmy is not common in this species. In species where biparental inheritance is known to be common, the potential for heteroplasmy should be taken into account during experimental design and interpretation of results.
Experimental design considerations for CallHap analyses—When designing an experiment to feed into the CallHap pipeline, consideration must be given to (1) the spatial scale of sampling, (2) the size of pooled libraries, (3) the choice of an appropriate reference genome for sequence alignment and variant discovery, and (4) the minimum read depth used. Each of these are discussed in turn below.
Fig. 6.
Haplotypes solution for Lasthenia californica de novo alignment. (A) Consolidated network phylogeny for CallHap solutions with the lowest RSS value (0.003002). Black indicates starting haplotypes, blue indicates new haplotypes fixed in the best solutions from the initial haplotype calling run, and green indicates new haplotypes found in the second haplotype calling run. For the second run, node size is scaled to indicate the number of output solutions in which a new haplotype occurred. Hash marks indicate number of SNPs that change identity along a branch. (B) Regression plot for lowest-RSS value CallHap solutions.

Spatial scale of sampling—Experimental designs that produce data for the CallHap pipeline will differ primarily on the geographic scale of sampling. For this purpose, small-scale sampling (e.g., for population genetics) indicates that populations are sampled at distances smaller than the hypothesized average dispersal distance of the target species, and large-scale sampling (e.g., for phylogeography) indicates that populations are sampled at distances greater than the hypothesized average dispersal distance. In population genetic studies, we expect that genetic structure is governed by gene flow and genetic drift such that all haplotypes have a reasonable chance of being sampled from any populations. At larger spatial scales, mutation rates exceed gene flow such that different regions may be characterized by different groups of closely related haplotypes.
At small scales, dispersal is great enough that each haplotype may be found in any location. Because of this, populations are differentiated primarily by differences in the frequencies of shared haplotypes, meaning that experiments should be designed with one SSL and one PL per population. In this type of experiment, there is a lowered likelihood of difficulties in recovering the correct haplotype network phylogeny and frequencies. At large scales, populations in close proximity to each other may represent a unique clade of related haplotypes. As shown in the test networks, it becomes difficult to place new haplotypes within clades when only one SSL is available for each clade. Additionally, if a haplotype is only present in a single population (pool), it is difficult to accurately place the haplotype within the network phylogeny. For large-scale studies, it would be advisable to create artificial pools by pooling samples from individuals from across the entire range. Notably, these pools should not include the samples used for SSLs. In addition, artificial pools should be constructed to consist of each sample at a different concentration in order to resolve the frequencies of SNP alleles that are shared among haplotypes, which will allow for a more robust inference of the phylogeny. Sequencing more than one SSL per population should also be considered in these cases. Sequencing multiple SSLs per region combined with artificial PLs will help resolve topologies and haplotype frequencies when the distance among populations within each region occurs at a small scale and sampled regions occur at a large scale.
Table 2.
RSS values and residual statistics for Lasthenia californica calculated by SNPs. For the sample size of 20 individuals per population, the 5% frequency separating estimates of the number of individuals carrying a haplotype is approximately the same as a squared residual value of 0.0025.

One final complication is that the true scale of a project may not become evident until after completing data analysis. For example, when the L. californica experiment was designed, the hypothesized dispersal range was greater than the distance among populations. However, after sequencing, we realized that seed dispersal in L. californica is much more limited than anticipated. In retrospect, creating artificial pools to help resolve the haplotype network phylogeny would have facilitated the estimation of the network phylogeny and haplotype frequencies.
Pooling and pooled library size—Many Pool-Seq protocols combine samples before DNA extraction (Kofler et al., 2012; Martins et al., 2014; Bélanger et al., 2016), but this will generate higher errors in SNP frequencies because equal amounts of tissue may not contain equal amounts of genomic DNA. In contrast, data for use in CallHap should come from libraries where DNA is extracted before being pooled to ensure equimolar proportions of DNA from each individual. Although populations of any size could be analyzed, sequencing error, pipette volume, DNA concentration, and consideration of sequencing limitations (see below) limit the number of individuals that can be placed in a single pool and still give accurate resolution of haplotype frequencies. On the other hand, if too few individuals per population are used, some haplotypes present in the population may be missed.
Table 3.
RSS values and residual statistics for Lasthenia californica calculated by population. For the sample size of 20 individuals per population, the 5% frequency separating estimates of the number of individuals carrying a haplotype is approximately the same as a squared residual value of 0.0025.

We can use the pool SNP frequencies from the L. californica study to estimate the error, which will provide a guideline for the maximum pool size. Examination of deviations from the expected values of the nearest multiple of 5% (i.e., for a pool size of 20) reveals an average error of 0.37% with more than 70% of deviations less than 0.25%, and only 5% greater than 2.0%. This error is very small and indicates that well over 200 samples could be included in each pool. Although a large number of individuals per population could be used to average out differences in cpDNA relative to total genomic DNA and experimenter error, there will be diminishing returns due to the resources required to isolate and quantify DNA from larger numbers of samples per pool, and fewer libraries can be multiplexed for capture and sequencing (see the section on read depth). In the L. californica study, a sample size of 20 individuals per population was used; this number provided reasonable accuracy in SNP frequency estimates while still capturing adequate haplotype diversity present in populations.
Choosing a reference genome—CallHap assumes that SNPs detected by variant calling arise from closely related haplotypes. Because of this, the CallHap pipeline requires that all libraries be aligned to a single reference genome. Because the genome used will have a large influence on the number and quality of SNPs generated, genome selection is an important aspect of any study using CallHap. In choosing a reference genome to use for CallHap analysis, preference should be given to conspecific genomes. If no such reference exists, one library of whole-genome shotgun sequencing (i.e., not subjected to targeted capture) should be included in the Illumina multiplex for de novo genome assembly. Although a de novo genome can be created using captured cpDNA, based on our experience, the incomplete nature of the capture makes it more difficult to carry out the de novo assembly. If creating a de novo reference is infeasible, adequate SNP calling can be conducted using a more distantly related reference. Limitations of interspecific references include the addition of artificial SNPs introduced due to alignment ambiguities that may be caused by fixed differences between the chloroplast genomes of the two species.
Minimum read depth—Another important parameter to consider when analyzing sequence data is the minimum read depth required to confidently identify genomic variants. To determine the minimum depth for the Lasthenia data, we ran the VCF filter multiple times with different depths and counted the number of unique haplotypes obtained each time. In general, minimum depth should be no less than 15 times the number of individuals in a pool (Sims et al., 2014), which would be 300 for a pool size of 20 individuals. Note that pooling larger numbers of individuals will require greater read depth and will induce limits on the number of SSLs and PLs that can be multiplexed for sequencing. For robust haplotype estimation, we suggest increasing the read depth until the number of haplotypes starts to decrease substantially (Fig. 7). We found that the optimum read depth value changes depending on the peculiarities of different species and sequencing runs; for L. californica, the optimum read depth was around 600, whereas for a separate study with Ranunculus occidentalis Nutt., the optimum minimum depth was found to be about 400 reads (J. Persinger et al., unpublished data).
Fig. 7.
Depth analysis for Lasthenia californica. The number of unique single-sample library haplotypes shows a substantial decrease at around 600 depth.

Analysis of CallHap outputs—Methods used for analysis of haplotype frequency data from CallHap will vary depending on the goals of the study. Population genetic studies utilizing nuclear genetic markers in diploid organisms typically use Wright's FST (Wright, 1949) or a similar analogue (GST, G′ST, DST, etc.; Whitlock, 2011). However, FST is based on comparisons of observed and expected heterozygosity at different scales and, consequently, is inappropriate for use with haplotype data. Instead, genetic distance measures that allow for variable ploidies and number of alleles per locus, and are not reliant on heterozygosity, such as Nei's genetic distance (Nei's D; Nei, 1973), Cavalli-Sforza and Edwards' chord distance (Cavalli-Sforza and Edwards, 1967; Edwards, 1971; Hartl et al., 1997), Φ-statistics (Meirmans, 2006), or haplotype genetic diversity measures (e.g., unbiased haplotype diversity; Gardner et al., 2015), should be used.
Methods such as Nei's D rely on calculations of the probability that the same combination of alleles will be found in two different populations; consequently, such methods are more appropriate for small-scale studies. When no haplotypes are shared between two populations, Nei's D gives an infinite distance between those populations; such a pattern indicates that dispersal rates among the populations sampled are very low, and that the accumulation of local mutations is the primary factor contributing to the genetic structure of populations. Limited dispersal relative to the scale of sampling will lead to haplotypes within populations being more closely related than to haplotypes in different populations. In these cases, methods such as chord distance or Φ-statistics may be more appropriate.
When genetic structure is governed primarily by limited dispersal leading to limited sharing of haplotypes across the sampled region, phylogeographic methods (Templeton, 1998, 2009; but see Knowles, 2008) or other methods of statistical phylogeography (Nielsen and Beaumont, 2009; Csilléry et al., 2010) should be used. These methods explain distributions of genetic variation using statistical inference and simulations of population history events by comparing observed data to different modeled population histories.
Applications—The CallHap pipeline has the potential to create a range of new opportunities for studies of cpDNA population genetic structure and phylogeography. This method provides accurate and economical estimates of seed-mediated gene flow by allowing for the use of pooled population sequencing data for cpDNA. Data for use in the CallHap pipeline come from population-level sampling of haploid genomes, including plant chloroplast genomes, mitochondrial genomes, and prokaryotic bacterial genomes. Because CallHap assumes all generated haplotypes are closely related and requires that all libraries examined be aligned to a single reference genome, this protocol should not be used for microbiome and microbial community studies. Outputs generated by CallHap can be analyzed using a variety of methods, including Nei's genetic distance, Cavalli-Sforza and Edwards' chord distance, Φ-statistics, and a variety of phylogeographic analysis methods in statistical phylogeography. The CallHap program, along with sample data and output files, is available at https://github.com/cruzan-lab/CallHap.
ACKNOWLEDGMENTS
The authors thank Tina Arredondo, Monica Grasty, Jamie Schwoch, and Pamela Thompson for help with sampling, DNA extraction, and sequencing library preparation. This research was supported with funds from a National Science Foundation Macrosystems Biology award (no. 1340746).