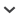We define a genetic species as a group of genetically compatible interbreeding natural populations that is genetically isolated from other such groups. This focus on genetic isolation rather than reproductive isolation distinguishes the Genetic Species Concept from the Biological Species Concept. Recognition of species that are genetically isolated (but not reproductively isolated) results in an enhanced understanding of biodiversity and the nature of speciation as well as speciation-based issues and evolution of mammals. We review criteria and methods for recognizing species of mammals and explore a theoretical scenario, the Bateson–Dobzhansky–Muller (BDM) model, for understanding and predicting genetic diversity and speciation in mammals. If the BDM model is operating in mammals, then genetically defined phylogroups would be predicted to occur within species defined by morphology, and phylogroups experiencing stabilizing selection will evolve genetic isolation without concomitant morphological diversification. Such species will be undetectable using classical skin and skull morphology (Morphological Species Concept). Using cytochrome-b data from sister species of mammals recognized by classical morphological studies, we estimated the number of phylogroups that exist within mammalian species and hypothesize that there will be >2,000 currently unrecognized species of mammals. Such an underestimation significantly affects conclusions on the nature of speciation in mammals, barriers associated with evolution of genetic isolation, estimates of biodiversity, design of conservation initiatives, zoonoses, and so on. A paradigm shift relative to this and other speciation-based issues will be needed. Data that will be effective in detecting these “morphologically cryptic genetic species” are genetic, especially DNA-sequence data. Application of the Genetic Species Concept uses genetic data from mitochondrial and nuclear genomes to identify species and species boundaries, the extent to which the integrity of the gene pool is protected, nature of hybridization (if present), and introgression. Genetic data are unique in understanding species because the use of genetic data 1) can quantify genetic divergence from different aspects of the genome (mitochondrial and nuclear genes, protein coding genes, regulatory genes, mobile DNA, microsatellites, chromosomal rearrangements, heterochromatin, etc.); 2) can provide divergence values that increase with time, providing an estimate of time since divergence; 3) can provide a population genetics perspective; 4) is less subject to convergence and parallelism relative to other sets of characters; 5) can identify monophyly, sister taxa, and presence or absence of introgression; and 6) can accurately identify hybrid individuals (kinship and source of hybrid individuals, F1s, backcrosses, direction of hybridization, and in concert with other data identify which hybrids are sterile or fertile). The proposed definition of the Genetic Species Concept is more compatible with a description of biodiversity of mammals than is “reproductively isolated species.” Genetic profiles of mammalian species will result in a genetic description of species and mammalian diversity, and such studies are being accelerated by technological advances that reduce cost and increase speed and efficiency of generating genetic data. We propose that this genetic revolution remain museum- and voucher specimen–based and that new names are based on a holotype (including associated tissues) deposited in an accredited museum.
Taxonomic and systematic literature pertaining to issues concerning species, speciation models, and whether species even exist in nature is voluminous. Even if one restricts the review to vertebrates and narrows the topics to species concepts and how to theoretically recognize species, the literature is impressive (e.g., Claridge et al. 1997; Coyne and Orr 2004; Howard and Berlocher 1998; Wheeler and Meier 2000). Further, the definition of “species” includes remarkable disparity concerning complexity, criteria, and application. For example, 4 definitions include phraseology such as “reproductively isolated from other such groups” (Mayr 1942:120), “tokogenetic entities that function in the phylogenetic system” (Wiley and Mayden 2000:74), “the smallest aggregation … diagnosable by a unique combination of character states” (Wheeler and Platnick 2000:58), and “smallest monophyletic groups deemed worthy of formal recognition” (Mishler and Theriot 2000:47). Employing such variation and associated criteria in an application of “species concepts” would clearly alter the number of recognized species.
Complexity of species definitions is not new; the 1st volume of the Journal of Mammalogy contained evidence of philosophical differences between Merriam (1919) and Taverner (1920) on how to delineate species of mammals. Merriam opposed efforts to invoke the “criterion of intergradations” as a measure of hybridization between morphological types, whereas Taverner (1920:126) stated “the species is a definite entity and its essential quality is its genetic isolation.” Merriam's (1919:7) position was typological and interpreted by Engstrom et al. (1994:181) as a defense of the Morphological Species Concept. Engstrom et al. (1994) proposed Taverner's (1920) position as recognition for the Biological Species Concept. However, we interpret Taverner's position as more likely to embrace the Genetic Species Concept (Bateson 1909) than the Biological Species Concept. This difference in opinion by mammalogists, at the time of the origin of the American Society of Mammalogists, indicates that species recognition has been a protracted evaluation of species models and their applications. As noted by Engstrom et al. (1994), Merriam's species concept resulted in recognition of 2 genera and 78 species of brown bears (Ursus arctos) that currently (Hall 1984) are recognized as a single species. Interestingly, a single species of brown bear originally was advanced by Allen (1876) in his seminal work on intraspecific geographic variability in mammals.
Mammalogists, for the most part, have avoided debate on species concepts, but most investigators follow a particular species definition. Most catalogues and checklists of species, such as Wilson and Reeder (1993, 2005), generally are not constructed under a single species concept but reflect diversity in philosophy of individual authors. Consequently, they appear to be accepted with little reference or adherence to species definitions and with little concern for conceptual standards (species concepts). Historically, there has been moderate to strong support for the Biological Species Concept among mammalogists (Mayr 1942). However, there has been a trend in recent issues of Journal of Mammalogy toward application of the Phylogenetic Species Concept defining monophyletic clades in phylogenetic trees identified by shared derived character states as species. Reasons that the Biological Species Concept has been accepted widely among mammalogists include the fact that all mammals are sexually reproducing and most are diploid (polyploidy is rare—Gallardo et al. 1999; Svartman et al. 2005). Further, there is an historical acceptance that a certain level of morphological difference indicates 2 reproductively isolated species (Corbet 1997), which is the basis for recognition of most allopatrically distributed species of mammals.
Before 1985 and the onset of DNA data sets, most descriptions of extant species of mammals were based on morphological analyses of museum voucher specimens and their associated databases. As early as the 1960s, other data sets (chromosomes, allozymes, scanning electron microscopy, etc.) to discriminate species were available, but they were championed by few mammalian systematists because they often required extensive expertise, expensive equipment, specific types of samples (in vitro incubated bone marrow, specially fixed tissues, etc.), or even laboratory colonies not available to the systematics community at large. Further, most mammalogists concerned with recognition of species and their relationship to other Linnaean taxa were affiliated with natural history museums. This affiliation allowed individuals to share comparative material and permitted systematists to examine closely related species across geographic ranges of taxa under study. Therefore, in almost all cases, the most comprehensive data set available for comparison of potential species was voucher specimens housed in natural history museums.
It is possible that the study of voucher specimens using classical morphology has resulted in a complete and accurate list of species of mammals and that more modern molecular data sets will not reveal many additional species or redefined species boundaries from those recognized in Wilson and Reeder (1993, 2005). However, we argue that there is substantial evidence demonstrating that this is not true.
Genetic Data and Species Recognition in Mammals
Methodological advances in molecular biology have led to generation of several genetic-based data sources. These included karyotypic (Hsu 1979; Patton 1967) and starch-gel allozymic data (Hubby and Lewontin 1966; Lewontin and Hubby 1966) developed in the mid-1960s and DNA-sequence data in the mid-1980s. Examination of these data sets demonstrates that there are cryptic species of mammals that would likely not be recognized based solely on classical studies of morphology of voucher specimens housed in museums.
Cryptic species of mammals identified by karyotypes and allozymes
There are several examples of mammals that are sympatric and behave as separate species that probably would not have been recognized without data from karyotypes and allozymes. These include Thomomys bottae–umbrinus (Patton and Dingman 1968), Sigmodon arizonae–hispidus–mascotensis (Zimmerman 1970), Macrotus californicus–waterhousii (Davis and Baker 1974), Mus domesticus–musculus (Capanna et al. 1976), Rhogeessa genowaysi–tumida (Baker 1984), Lasiurus ega–xanthinus (Baker et al. 1984), Sorex araneus–coronatus–granaries (Hausser et al. 1985; Searle 1988), and Peromyscus beatae–boylii–levipes (Houseal et al. 1987; Schmidly et al. 1988). In those examples and others noted in Corbet (1997), differences that distinguish 2 or more species were chromosomal and allozymic. Many of those species were sympatric, with no karyotypic or allozymic evidence of hybridization. If hybridization was present, most hybrids were less fertile than nonhybrids and introgression was restricted to a narrow zone, indicating that genetic isolating mechanisms had evolved. Consequently, there was justification for recognition of 2 or more species. Although chromosomal and allozymic data are useful in systematics, neither method provides as much resolution as DNA-sequence data nor are they used as frequently today as are DNA-sequence methods.
Cryptic species of mammals identified by DNA sequences
Over the last 2 decades, a broad-based DNA-sequence data set has become available to examine biodiversity and speciation in mammals. This data set is a product of methodological advances associated with automated DNA sequencing, polymerase chain reaction, universal primers, GenBank, human genome project, biological informatics, software to analyze such data (reviewed 3 February 2006 at http://evolution.genetics.washington.edu/phylip/software.html), and genetic resource collections associated with voucher specimens. DNA sequencing is available at nearly all research and educational institutions and with GenBank and biological informatics (Baker et al. 1998), studies of local populations or favorite species are possible for many researchers even at smaller institutions with limited budgets and no museum collections.
Initial results from DNA-sequence databases indicate that cryptic species are more common in mammals than previously thought. Some species that have been recognized and described or justified using DNA-sequence data include Loxodonta cyclotis (Roca et al. 2001), Carollia sowelli (Baker et al. 2002), Neotoma macrotis (Matocq 2002), Myotis occultus (Piaggio et al. 2002), Notiosorex cockrumi (Baker et al. 2003), Lophostoma equatorialis (Baker et al. 2004), Peromyscus schmidlyi (Bradley et al. 2004a), Reithrodontomys bakeri (Bradley et al. 2004b), Lagidium boxi (Spotorno et al. 2004a), and Thylamys cinderella (Braun et al. 2005).
To summarize, there is evidence that species of mammals are unrecognized, and a database of DNA sequences for most mammalian species is now attainable, financially feasible, and rapidly being compiled. It is our position that the DNA database permits substantially more resolution to understand species presence and boundaries than any previously available database. The DNA database permits application of the Genetic Species Concept to understand mammalian biodiversity. Although scientific breakthroughs to generate the DNA database are relatively recent (last 25 years), the Genetic Species Concept (Bateson 1909) is not new, and along with the Typological and Biological Species Concept (Dobzhansky 1937; Mayr 1942), is among the oldest species concepts.
Genetic Species Concept
There are multiple definitions and descriptions of what could be interpreted as a Genetic Species Concept (Avise and Ball 1990; Baker and Bickham 1986; Bateson 1909; Butlin 2005; Dobzhansky 1950; Masters and Spencer 1989; Mayden 1997; Mayr 1969; Muller 1939; Nei 1976; Schilthuizen 2000; Simpson 1943; Taverner 1920). Most of the ideas that relate to speciation models (including the Genetic Species Concept) have been hashed, rehashed, and tweaked in the literature, and it is not our goal to establish a new concept. It is our goal, however, to organize ideas from the published literature into a pragmatic perspective to explore patterns of genetic variation in mammalian taxa and to accurately identify species and species boundaries to better understand mammalian systematics, evolution, and biodiversity. This perspective is intimately linked to the power of resolution that DNA-sequence and molecular data provide to document species boundaries, hybridization, monophyly, introgression, and so on.
We define genetic species as a group of genetically compatible interbreeding natural populations that is genetically isolated from other such groups. Under our definition of the Genetic Species Concept, speciation is the accumulation of genetic changes in 2 lineages (Bateson 1909) that produce genetic isolation and protection of the integrity of the 2 respective gene pools that have independent evolutionary fates. Therefore, the process of speciation depends on divergence in genes, the genome, and chromosome structure (Check 2005), and species exist when the integrity of 2 gene pools is protected as a consequence of genetic differences in their respective genomes (e.g., as outlined in the Bateson–Dobzhansky–Muller [BDM] model but not restricted to those conditions).
A comparison of our definition of the Genetic Species Concept to the Biological Species Concept, Morphological Species Concept, and Phylogenetic Species Concept is presented in Table 1. How does our definition of the Genetic Species Concept differ from the Biological Species Concept? Under the Biological Species Concept, the definition is “a group of interbreeding natural populations that is reproductively isolated from other such groups” (Mayr 1942:120). In contrast, genetic isolation is the primary feature of the Genetic Species Concept. Because members of interbreeding populations that do not have reproductive or genetic isolation are recognized as conspecifics and because populations that are reproductively and genetically isolated are recognized as distinct species, these definitions overlap between the 2 concepts. Where the 2 concepts differ is when genetic isolation exists but reproductive isolation does not. In this special case, the Biological Species Concept (sensu Dobzhansky [1937] and Mayr [1942], but see the definition of Coyne and Orr [2004] in chapter 1) would recognize the former as conspecific but the Genetic Species Concept would recognize 2 different species. Although we know of no example in mammals where reproductive isolation has evolved without genetic isolation, it is possible for genetic isolation to evolve without reproductive isolation (Table 2). There also are differences in levels of interbreeding (hybridization) between 2 genetic species. These can range from production of no hybrids to all sterile hybrids, to a hybrid zone (or zones) that have all combinations of possible crosses with all individuals fertile and with introgression being trivial beyond that zone. In cases where the integrity of the 2 gene pools is maintained, it is our position that these gene pools represent genetic species. Clearly, examples with extensive hybridization with fertile hybrids do not fit the definition “species are reproductively isolated from each other” (Mayr 1942:120). We predict that when genetic profiles become available for populations currently recognized as conspecifics (Wilson and Reeder 2005), numerous examples of genetically isolated populations that are not reproductively isolated will be identified. Recognizing these examples as genetic species will be the best means to describe biodiversity in mammals.
The 1st reported example in mammals that we know of where genetic isolation was documented but reproductive isolation was not present was a study of pocket gophers of the genus Thomomys (Patton and Dingman 1968). To justify recognition of species status despite absence of reproductive isolation, Patton and Dingman (1968:11) stated that recognition “of the 2 forms as distinct species is in greater accord with the biological inferences. This interpretation allows for a greater appreciation and understanding of the past historical events [and] present distribution.” We further develop the idea that recognition of these “genetic species” provides more information for understanding biodiversity than relegating them to subspecific status.
Species (Table 2) that are not reproductively isolated but appear to be genetically isolated include mule deer and white-tailed deer (Odocoileus hemionus–virginianus), pocket gophers (Thomomys bottae–townsendii and Geomys bursarius–knoxjonesi), African elephants (Loxodonta africana–cyclotis), and tent-making bats (Uroderma bilobatum–davisi). The final example, Peromyscus leucopus–texanus, has a level of hybridization that may or may not merit recognition of specific status but the point is that the hybrid zones will display varying degrees of hybridization, introgression, and so on, and each will need to be evaluated on its own genetic structure. In these examples, considerable reproduction occurs between individuals of different species, including fertility of F1 and backcross individuals (Table 2), yet 4 of these examples are recognized as species in Wilson and Reeder (2005). We propose that these examples of hybridization between species pairs more closely fit our definition of the Genetic Species Concept than they do a concept focused solely on reproductively isolated populations. The extent of hybridization in mammals that will be revealed by examination of genetic data from future studies will document numerous examples that fit the definition of the Genetic Species Concept proposed herein.
Alternatively, one could argue, as have Coyne and Orr (2004), that the Genetic Species Concept is equivalent to the Biological Species Concept or that the Genetic Species Concept is a subset of the Biological Species Concept. Coyne and Orr (2004) redefined the Biological Species Concept to allow hybridization and some reproduction between 2 species and extensively discussed isolating mechanisms that would permit such hybridization.
For the most part, we agree with the interpretations of Coyne and Orr (2004), and their additions of hybridization to the Biological Species Concept overlaps with our definition of the Genetic Species Concept. However, we interpret reproductive isolation as invoked by Mayr (1942) as the end point in a series of genetically based events. Mayr's perspective of genetics and speciation is reviewed by Provine (2004:1045), and he concluded that Mayr thought speciation “boiled down to the genetics of natural populations,” which is compatible with our position; however, Mayr's focus was on reproductive isolation rather than genetic isolation. A classification of reproductive isolating barriers is presented by Coyne and Orr (2004:28) and Avise (2004:324).
Under the Genetic Species Concept, genetic isolation results as 2 genomes diverge to the point that they are genetically distinct and no longer share a common evolutionary fate. There are multiple ways that genetic isolation can result in premating or postmating mechanisms. These include, but are not limited to, sequence divergence in genes that are not functional in combination with genotypes in the sister species (reviewed in Coyne and Orr 2004); cytoplasmic–nuclear incompatibility (Asmussen et al. 1987); genetic changes that lead to behavioral changes or changes in pheromones and odors associated with conspecific recognition (Wickliffe et al. 2003); chromosomal rearrangements that produce infertile or less-fertile hybrids (Baker and Bickham 1986; White 1978); genetic changes that alter timing or levels of gene expression; and disruption of coadaptive gene complexes (Shaw 1996).
The distinction between “genetically isolated and reproductively isolated” and “genetically isolated but not reproductively isolated” is significant to understanding many evolutionary processes, including speciation. If, as described above (Coyne and Orr 2004), the Biological Species Concept is redefined to permit extensive hybridization between species, it is critical to the study of mechanisms and processes involved in evolution of protection of the integrity of gene pools to distinguish cohorts of species that are reproductively and genetically isolated from those that are only genetically isolated, especially when there is extensive fertility among hybrid individuals (Table 2). Studies addressing mechanisms of speciation in animals have focused on Drosophila (reviewed by Coyne and Orr 2004). The ability to sequence entire genomes and the variety of model systems in natural species of mammals where hybridization occurs but the integrity of the gene pool is protected (Table 2) permits examination of speciation in mammals using methods such as those in chapter 2 of Coyne and Orr (2004).
We hypothesize that hybridization will be common among genetic species of mammals and to imply that they are “reproductively isolated” is inaccurate. By our definition, these 2 phylogroups are genetic species if the integrity of the gene pools of the 2 phylogroups is protected. Unless there is a redefinition of “reproductive” that states that reproductive isolation equals genetic isolation in the presence of hybrids, or alternatively, if the definition of the Biological Species Concept becomes “a group of interbreeding natural populations that is reproductively and/or genetically isolated from other such groups,” there is an incompatibility in the dictionary definition and the commonly understood usage of the phrase “reproductive isolation” and the species status proposed for the Biological Species Concept by Coyne and Orr (2004). After all, it is the Biological Species Concept we are discussing, not the Reproductive Species Concept, and adding the word “genetically” to more accurately describe speciation as envisioned in Coyne and Orr (2004) and further developed herein is a more accurate description of speciation in mammals.
How does our definition of the Genetic Species Concept differ from the Phylogenetic Species Concept? There are several different definitions of the Phylogenetic Species Concept, but one definition is “the smallest population or group of populations within which there is a parental pattern of ancestry and descent and which is diagnosable by unique combinations of character states” (Cracraft 1997:329). Although this is a popular and useful definition, its application does not usually include evidence of separate evolutionary fates or isolation. Although there is overlap in the application of the Genetic Species Concept and Phylogenetic Species Concept in use of monophyly and sister-species status, in the application of the Genetic Species Concept proposed herein, there is greater emphasis on supporting data for isolation and proof that there is protection of the integrity of the gene pool.
We chose to use “Genetic Species Concept” because we think this definition better describes variation present in nature. Our definition is built upon 1) genetic speciation, 2) genetic definition of species, 3) genetically defined phylogroups, 4) evidence of protection or integrity of gene pools in the presence of hybridization, 5) significance of genetic differentiation in phylogroups that are not morphologically distinct, 6) the way genetic data offer better resolution than any other systematic database, 7) the way breakthroughs in genetic methods will result in DNA profiles that will be used to define species and species boundaries, and 8) application of genetic data. Our position is that studies of these phylogroups through molecular methods will provide an opportunity to understand evolution of isolating mechanisms, role of ecological features in speciation, and many other poorly understood evolutionary phenomena. Our position is best described by Brookfield (2002:107: not seen, cited in Coyne and Orr 2004:25):
The essence of the “species problem” is the fact that, while many different authorities have very different ideas of what species are, there is no set of experiments or observations that can be imagined that can resolve which of these views is the right one. This being so, the “species problem” is not a scientific problem at all, merely one about choosing and consistently applying a convention about how we use a word. So, we should settle on our favorite definition, use it, and get on with the science.
Resolving power of the Genetic Species Concept
The Genetic Species Concept provides a unique level of resolution for systematists to study the number of species and species boundaries. Toward this resolution, use of genetic data 1) can quantify genetic divergence from many different aspects of the genome (mitochondrial and nuclear genes, protein-coding genes, regulatory genes, mobile DNA, microsatellites, chromosomal rearrangements, heterochromatin, etc.); 2) can provide divergence values that have increased with time providing an estimate of time since divergence; 3) can provide a population genetics perspective of the magnitude and types of divergence; 4) is less subject to convergence and parallelism than use of any other set of characters; 5) can accurately identify monophyly and sister taxa; and 6) can accurately identify hybrid individuals (kinship and source of hybrid individuals, F1s, backcrosses, extent of introgression, and direction of hybridization), and in concert with use of other data can provide an estimate of the level of fertility of F1 hybrids and subsequent hybrid crosses.
Other conceptual considerations
In addition to genetic versus reproductive isolation, there are some other points supporting the uniqueness of a Genetic Species Concept. First, there are additional implications from the Genetic Species Concept as related to other concepts, such as the Evolutionary Species Concept (Simpson 1961). For example, the definition of Wiley (1978:18) or Wiley and Mayden (2000) of “a single lineage of ancestor–descendant populations which maintains its identity from other such lineages and which has its own evolutionary tendencies and historical fate,” does not allow for recognition of speciation over time within an ancestor–descendant lineage as would be predicted to occur with adequate time for such genetic divergence to evolve.
Second, the so-called Barcode Species Initiative (Hebert and Gregory 2005), that a few hundred base pairs from a single gene from a few individuals can identify presence or absence of a species, has been highly criticized (Ferguson 2002; Moritz and Cicero 2004; Roca et al. 2005) and is not the Genetic Species Concept embraced in this paper (although viewing genetic divergence in a single gene is the 1st step in our application). Conceptually, the Barcode Initiative cannot resolve presence–absence of species or the extent of isolation between 2 mitochondrial gene modifications without additional data to understand inter- and intraspecific variation within and between species. Further, sole use of a mitochondrial gene does not resolve questions about hybridization, gene flow, and introgression. With a proper genetic profile for all the species of concern, it will be possible to use a single metric to identify species; to determine boundaries of species and intraspecific variation and to insure the accuracy of implementation for the Barcode Initiative. We propose that such a genetic profile is the most critical step in the application of the Genetic Species Concept.
Genetic Species Concept, the Bateson–Dobzhansky–Muller Model, and Speciation in Mammals
Part of the theoretical basis for the Genetic Species Concept is embodied in the BDM model that was 1st outlined nearly a century ago by Bateson (1909) and was further defined by Dobzhansky (1934) and Muller (1939). In the BDM model (Gavrilets 2003), accumulation of genetic changes in 2 separate populations results in 2 species. The following from Coyne and Orr (2004:269) explains the process:
Consider two allopatric populations that evolve independently. Each experiences many substitutions over long periods of time until the populations become distinct genetically. If we could take a diverged gene from one population and place it in the genome of the other, would it work? It is easy to imagine that the gene might be reasonably effective on this related genetic background. It is also easy to imagine that it would not work well. But it is hard to imagine that it would often work better than on its own genetic background. This simple asymmetry forms the basis of Dobzhansky and Muller's model. Genes within a population are selected to work well together while genes from different populations have not been tested together. On average, then, we expect a mixture of genes from two species to be less well adjusted than those from a single species. Hybrid sterility or invariability might therefore be a simple byproduct of the divergence of genomes that are geographically isolated. Nowhere do we have to suggest that natural selection opposed any step in this process.
What is the relevance of this model to understanding speciation in mammals? To answer this question, it is necessary to know, if the BDM model is operative in mammals, what kind of patterns of genetic divergence and genetic isolation would be expected. It is well documented that even in stable geologic times, allopatric populations are present in most mammalian species, and gene flow between or among these populations is not possible. Barriers producing allopatry are sometimes obvious, such as water between islands and mainlands, mountains between lowlands, forests between grasslands, grasslands between forests, and so on. Other situations that potentially result in allopatric populations are climatic changes associated with cycles of ice ages and interglacial periods that changed distributions of habitat types for long periods of time. Assuming these periods of allopatry were of sufficient time for populations to accumulate genetic changes that produce isolation, the BDM model would produce genetic species through normal genetic divergence over time (see the works of Fitzpatrick [2004] for why reproductive isolation may be more rapid in mammals than in birds or other vertebrates, and Gavrilets [2003] for mathematical models of relationships of factors affecting evolution of genetic isolation). If during these cycles of allopatry, selective conditions were stabilizing on allopatric populations that were conspecific at the beginning of allopatry, then it would be expected that genetic isolation would be accompanied by minimal morphological evolution and divergence. The examples in the sections described above in “Cryptic species of mammals …” fit predictions of evolution of species through the BDM model under stabilizing selection. If ≥1 of the allopatric populations experienced different directional selection or if sufficient genetic drift was present, then morphologically distinct species would be expected. This would include most of the closely related and morphologically poorly defined species recognized in Wilson and Reeder (2005).
We hypothesize that speciation by the BDM model is the primary means of genetic isolation for many mammalian species. Such genetic isolation often will not be accompanied by morphological divergence at the level that is typically thought to indicate reproductive isolation in allopatric populations (Corbet 1997). Within mammals, there is a pattern of morphological similarity or identity between closely related species where the primary distinction is size and other minor differences in morphological traits. For example, most species of Peromyscus essentially look alike. Further, the complexity of identifying species using morphology without an understanding of geographic or locality information is problematic. For species of Peromyscus, Myotis, Sorex, and Crocidura, it is difficult to develop keys that distinguish each species from its congeners based solely on morphology. We hypothesize that most speciation within these groups is a result of allopatrically isolated populations experiencing consequences outlined in the BDM model with resulting species either maintained or slightly altered by local selective forces or drift.
How common is the evolution of genetic changes within currently recognized conspecific groups that might contain unrecognized species? Avise and Walker (1999) noted that most species consist of ≥2 mitochondrial DNA (mtDNA) geographically defined, genetically distinct populations that are distributed much like that implied in typical subspecies maps in Hall (1981). Avise and Walker (1999) called these geographic subdivisions “phylogroups.” We interpret these genetically defined, wide-ranging geographic populations as suggesting a substantial time since a common ancestor was shared by the 2 or more phylogroups. During the last 5 years, we and others (Table 3) have used the cytochrome-b gene to provide a relative estimate of time since isolation for the pattern of phylogroups observed by Avise and Walker (1999).
How are maternal lineages typically distributed in mammalian species? Is most of the intraspecific genetic variation in the mitochondrial genome distributed within most local populations or is the major component of mtDNA variation distributed in geographically defined contiguous regions? We partitioned divergence values for the mitochondrial cytochrome-b gene into intrapopulational and intraspecific components (Table 3). Clearly, most populational studies for geographic and local variation in mtDNA haplotypes do not reveal a high level of intrapopulational variation but do reveal a high level of intraspecific variation that is geographically defined. These values are compatible with predictions of the BDM model of well-defined geographic phylogroups that result from genetic changes expected to accumulate during periods of allopatry. Distance values for populational, intraspecific, and sister species (Table 3) are essentially the same pattern of variation shown by Bradley and Baker (2001: figures 2, 4, 5, and 7).
How many phylogroups are present in mammalian species defined in Wilson and Reeder (1993). For this calculation, we used the species list in Wilson and Reeder (1993) because several of the conspecific phylogroups that were present in the 1993 edition are recognized as species in Wilson and Reeder (2005) based on our definition of the Genetic Species Concept. We outlined the frequency of such phylogroups in 61 species (>1%) of mammals (Table 4). Sister species of mammals that have been recognized as species based on morphology often have cytochrome-b distance values >5% and this magnitude of divergence in the cytochrome-b gene has been associated with taxa recognized as species (Bradley and Baker 2001). Phylogroups with a genetic divergence (distance) of >5% occurred in 32 of 61 species (Table 4), and several species as defined in Wilson and Reeder (1993) consisted of 3 phylogroups (8 species), 4 phylogroups (3 species), and 5 phylogroups (3 species). Within the 61 species, there were 55 phylogroups with values >5% divergence in the cytochrome-b gene (Table 4). This result is similar to that reported for mammals by Avise and Walker (1999: figure 2). If the frequency of phylogroups per species (55 phylogroups/61 species or 0.90 phylogroups/species) is a reasonable estimate for the total number of phylogroups in the remainder of the 4,629 species in Wilson and Reeder (1993), then there will be >4,100 such phylogroups in mammals. If 50% of these phylogroups have evolved sufficient nuclear divergence through the process of the BDM model to be genetically isolated from the other such groups within a so-called “species,” then the number of unrecognized species in Wilson and Reeder (1993) is >2,000.
There are several assumptions made in deriving an estimate of >2,000 currently unrecognized genetic species of mammals. First, we are assuming that the estimated number of phylogroups is likely characteristic of the unstudied 4,000+ species of mammals. For species such as Carollia brevicauda (n = 24), Glossophaga soricina (n = 38), Dermanura glauca (n = 35), and Uroderma bilobatum (n = 57), there is probably an adequate geographic sampling to estimate the number of phylogroups (Table 4). However, there are 16 species in Table 4 for which only ≤4 individuals have been sequenced. Such small samples may underestimate phylogroups. Second, essentially all species in Table 4 have been studied for intraspecific morphological variation before Wilson and Reeder (1993). We interpret this as meaning that this group of species is representative of taxa for which obvious morphological species would have been detected without DNA-sequence data. Many of the other 4,000+ mammalian species have been poorly studied and probably contain a greater percentage of unrecognized species than the taxa in Table 4. Third, species in Table 4 are rodents, bats, and shrews with small body sizes. At least theoretically, bats are much more vagile than are rodents and shrews. However, if large vagile mammals such as bovids, felids, elephants, and cetaceans have a much lower frequency of phylogroups than do small mammals, this would result in our overestimating the number of phylogroups. Most mammalian species, however, are rodents, bats, and shrews (70% of the 4,629 species in Wilson and Reeder [1993]). A substantial portion of the remaining 30% are not large, highly vagile mammals; if large, highly vagile mammals have significantly fewer phylogroups than the species in Table 4, there will be a reduction in this estimate, but the overview will remain that there is a substantial number of unrecognized species of mammals. Further, studies of African elephants (Loxodonta africana) suggest that they were distinguished by phylogroups indicating that not all large mammals are absent of genetically defined phylogroups (Roca et al. 2001).
The factor that could greatly alter the estimate of >2,000 unrecognized species is the assumption that 50% of these phylogroups represent species as defined by the application of the Genetic Species Concept. Certainly an insufficient number of species has been studied with a thorough genetic profile to adequately estimate how many of the phylogroups with a genetic divergence > 5% will result in data that documents complete genetic isolation. The choice of 50% for calculation is poorly defined statistically and will be better understood by genetic studies of other phylogroups with >5% divergence.
Genetic isolation is not simply an off-and-on switch because genetic changes in allopatric populations are accumulated slowly across the genome and may involve a substantial number (estimated at 200 for Drosophila—Presgraves 2003) of loci affecting isolation. Accumulation of adequate change in independent sister lineages that results in genetic isolation will be a chance event occurring rapidly in some cases but requiring long periods of separation in other cases. Genetic isolation resulting from the BDM model will be expected to produce intermediate and incomplete stages of reproductive isolation before the completion of reproductive isolation. We predict that genetic profiles of interactions between members of mammalian phylogroups will reveal examples of complete genetic isolation and examples of essentially no genetic isolation even with the same genetic divergence in the mitochondrial marker used to select phylogroups for more intensive study. But, more commonly in phylogroups with >5% genetic distance in the cytochrome-b gene, various combinations of genetic isolation will be apparent. As a result, when phylogroups are sympatric, there will often be hybridization, and data documenting the genetic basis for any level of isolation will be difficult to organize into well-defined stages (Table 2). Genetically defined hybrid zones will be common.
Before DNA-sequence data, the best-defined hybrid zones were the result of our recognizing chromosomal rearrangements, and there is extensive literature concerning gene flow and maintenance of such zones (Honeycutt and Yates 1994). Studies of hybrid zones and hybrids have provided insights into evolution of isolating mechanisms such as Kaneshiro's hypothesis (Bradley et al. 1991; Kaneshiro 1983; cf. Patton and Smith 1990), sexual differences in fertility (Haldane's rule—Coyne 1992), origin of evolutionary innovations (Arnold and Emms 1998; Butlin 1998), rare alleles (Bradley et al. 1993), and genome imprinting (Monk and Surani 1990). Hybrid zones in mammalian phylogroups defined by genetic changes predicted by the BDM model will permit experimental designs to study these and other evolutionary phenomena.
There are 5 significant conclusions from the above discussion. First, focus on genetic isolation rather than reproductive isolation results in a broader conceptual definition of a species. Second, if the BDM model is operating in mammals experiencing stabilizing selection, then genetically isolated populations will evolve that will not be easily detectable using classical studies of skin and skull morphology. Third, if the number of unrecognized cryptic species is anywhere near the above estimate of >2,000, there has been a significant underrepresentation of mammalian species; furthermore, that underestimation significantly affects conclusions on the nature of speciation in mammals, barriers associated with evolution of genetic isolation, estimates of biodiversity, designs of conservation initiatives, zoonoses, and so on. A paradigm shift relative to these and species-based issues will be needed. Fourth, the only type of data that will be effective in detecting these “genetic species” is genetic, especially DNA-sequence data. If this is true, it becomes important to develop methods to recognize morphologically cryptic genetic species and species boundaries. Fifth, it should be remembered that the above discussion is concerned primarily with speciation by the BDM model. There are other speciation modes and processes that result in undetected species of mammals. Any estimate of the number of unrecognized species of mammals would need to include consequences of speciation by these other processes.
Application of the Genetic Species Concept
Speciation is a genetic process and the status of a species is best decided with genetic data. Even in cases where the species are not distinguishable based on morphological features (cryptic species), such as Rhogeessa genowaysi/tumida, it can be easy to identify genetic species because they are sympatric with no hybrids (Baker 1984). Beyond such simple cases, the diverse nature of biological entities may cause difficulties in applying any species concept because examples are not clear cut (Corbet 1997, Hudson and Coyne 2002, Mayden 1997, Wiens and Penkrot 2002). What is new concerning application of genetic data is that the dataset can provide greater resolution to identify and understand hybridization and introgression, past and present (Jones et al. 1995). Decisions under the Genetic Species Concept will be based on larger and more complex datasets involving multiple mitochondrial and nuclear markers, which will be more resolving; however, in some cases there will be apparent incongruencies. Using such datasets to determine species will require an interpretive component and knowledge of molecular evolution. The following “application” is not presented in a rigid context but in the spirit of developing a working set of criteria to be used to discriminate species in light of natural genetic variability and tremendous amounts of data that will be detected by new sophisticated techniques.
We expanded the criteria of Bradley and Baker (2001) into a framework for interpreting significance of genetic divergence and resolving presence or absence of 2 genetic species. Specifically, we address issues of identifying unrecognized species, genetic variation associated with phylogroups, and sympatric versus allopatric populations.
Identification of populations with greatest probability of representing an unrecognized species
We used distance values for a protein-coding gene as evidence that populations have been isolated from each other for sufficient time to have speciated by the BDM model. For mammals, the cytochrome-b gene has proven to be an appropriate marker, but other markers (cytochrome oxidase I, etc.) also may be applicable. To establish a distance value that had a high probability of being associated with the completion of speciation, we used variation in distance values of cytochrome-b distinguishing sister species recognized as such by data other than genetic.
Populations that are distinguished by distance values equal to or greater than is typical of sister species listed in Wilson and Reeder (2005) merit further study using other molecular loci or methods to determine their stage of speciation. This step is the most misunderstood and criticized (Ferguson 2002; Mayden 1997; Will et al. 2005) of the effort to identify species through genetic distance because critics conclude that a certain distance value in a single gene will not always resolve the presence or absence of 2 species. We agree that the failure rate of a single metric (especially from the mitochondrial genome) to identify species may be high, although Hebert and Gregory (2005:857) claim a success rate of 96% for birds. Yet we also conclude that a single metric, such as the genetic distance of the cytochrome-b gene, when viewed against an appropriate database from other genetically defined species and subspecies with similar life histories and biological characteristics, will have an accuracy rate as great as any other single character used to identify species or populations for study.
In Bradley and Baker (2001), we conclude that a genetic distance < 2% was typical of populational and intraspecific variation, whereas values >10% usually were distributed in geographically discreet phylogroups typical of different biological species. It was not our intent to imply that a value >10% always indicates 2 biological species or that < 2% always identifies conspecific populations. Rather, we suggested that if the goal is to efficiently recognize cryptic or currently unrecognized genetic species of mammals, examples of phylogroups within a single species of mammal where the genetic divergence in the cytochrome-b gene is >10% will be the best group for study, and additional data (nuclear genes or morphological variation) should be collected to determine if an unrecognized species exist. Alternatively, if other phylogroups have a 2% divergence, then there is a lower probability of the presence of unrecognized species. A genetic distance of 5% will have a greater probability than 2% but less probability than 10%. In light of the above discussion, there are numerous examples where <5% distance value is present in sister species of mammals that had been recognized by morphological variation before study with molecular methods. A value of <5% will often identify unrecognized genetic species if data in Table 3 from sister taxa are predictive. Becoming genetically isolated by mutations at multiple loci as proposed in the BDM model will not be an ordered process during which a certain amount of time or genetic divergence in something as simple as the cytochrome-b gene will always yield the same percentage of genetic isolation for phylogroups under study. There is nothing unique or special about >5%; it is a subjective value chosen from the review of the published genetic distances for mammals. The value selected may be more predictive if variations in distance values are calculated by selected species cohorts with common generation time, metabolic rate, body size, and so on.
Populational and geographic variation of phylogroups
After 2 monophyletic populations with an appropriately high genetic divergence (>5%) are identified, the next step is to generate distance values from the cytochrome-b gene of multiple individuals from populations with sufficient geographic sampling to understand how pairwise distance values are distributed in the taxon under study. A major issue is to determine if haplotypes that are defined by a high level of genetic distance are distributed in geographically defined phylogroups and to resolve geographic limits of each phylogroup. At this point, it is necessary to determine if phylogroups under study are sympatric, parapatric, or allopatric. Application of the Genetic Species Concept for allopatric groups uses a different set of criteria and experimental design than for sympatric phylogroups.
Sympatric phylogroups
The experimental design to resolve specific status of sympatric phylogroups is to determine whether hybridization between the 2 phylogroups is present. If individuals of the phylogroups are sympatric and no hybrids are present, or if F1s are sterile, then 2 species exist. If F1s are fertile, then the extent of introgression needs to be determined. Extensive hybridization between 2 genetic species is permissible under some conditions, as described below.
References cited above in “Cryptic species of mammals identified by DNA sequences” are studies that address hybridization and introgression. This is a rapidly developing field that is linked closely to advances in biological informatics and genome sequencing. Much of the available software that can document sister taxon relationships, monophyly, hybridization (direction and extent), introgression, and unique genetic features is reviewed at http://evolution.genetics.washington.edu/phylip/software.html; last accessed 3 February 2006. A complete genetic profile for populations and species that may or may not be specifically distinct will provide more resolution than any other comparable data set. Detailed genetic profiles generated for “species” (Table 2) reveal that a complex of intermediate stages of genetic and reproductive isolation will be described and each of these will provide an opportunity to better understand the process of speciation.
Parapatric hybrid zones, integrity of the gene pool, and species status
Genetically distinct phylogroups (initially identified by variation in the cytochrome-b gene) are geographically distributed and will often have parapatric hybrid zones (Table 2). Concerning parapatrically distributed phylogroups, the criterion for species recognition versus intraspecific status is this: if in the narrow zone of contact no hybrids or only sterile hybrids are produced, then 2 species exist. Existing studies indicate that these genetically defined phylogroups often will have parapatric hybrid zones (Table 2). We propose that 2 phylogroups represent species when hybridization is restricted to a limited geographic area, and outside the hybrid zones respective phylogroups are defined by unique and concordant statistically supported monophyletic clades based on mitochondrial and nuclear genetic variation. What is crucial is protection of the integrity of the gene pool in that, although hybrid individuals are present, introgression beyond the hybrid zone is not significant. In this definition, the nature of hybridization (fertile F1s, backcross individuals, or whatever) is not critical. We interpret the unique genetic character states present in each respective phylogroup, beyond the hybrid zones, as evidence that genetic divergence that distinguishes phylogroups has evolved in the absence of significant genetic introgression from the other. Unless the hybrid zone is of recent origin, a narrow geographically restricted hybrid zone is evidence of genetic isolation and consequently both phylogroups have a high probability of an independent evolutionary fate. If respective members of these 2 phylogroups are morphologically unique from each other, the case for species status is further justified. The less well defined and greater the width of a hybrid zone and the higher the frequency of introgression in both mitochondrial and nuclear markers, the stronger the evidence is for justifying intraspecific variation as the proper classification for the significance of phylogroups under study. As detailed genetic profiles become available, considerable debate will be necessary to find an acceptable level of characteristics of hybrid zones and introgression that is permitted for genetic species as compared to variation appropriately assigned to a single species.
Some authors (Helbig et al. 2002:521–522) have referred to narrow hybrid zones as support for recognizing semispecies rather than species. Using the definition of Helbig et al. (2002), mule deer and white-tailed deer (Carr et al. 1986; Cathey et al. 1998) would be recognized as semispecies because there is historical introgression of the mitochondrial genome, multiple hybrid zones have been identified over a wide geographic area, and there is no evidence of loss of fertility in hybrid individuals. We reject the implementation of semispecies because, when compared to the proposed definitions above, the appreciation of the biodiversity in mammals is better described by assignment of specific status. It is significant that mule deer and white-tailed deer are recognized as distinct species based primarily on morphology, behavior, and ecological limits. Mule deer and white-tailed deer clearly have their own evolutionary fate and are adapted for specific ecological situations. From a biodiversity and conservation standpoint, it is most logical to recognize such entities as white-tailed and mule deer as species.
Allopatric phylogroups
Allopatric populations or phylogroups are problematic for application using most species concepts (allopatry is less a problem for some definitions of the Phylogenetic Species Concept that are not concerned with documentation of reproductive or genetic isolation of species). The most informed decisions for allopatric populations will be made by using the magnitude of genetic distance that typically is present in sister species for 1 or more genetic markers. Known sister species recognized from nonmolecular data sets must be studied to determine the range of genetic distance values for multiple loci that will serve as standards for estimating specific status of allopatric phylogroups. It would be ideal for sequence data sets generated for DNA motifs to contain multiple examples of sister-species comparisons to more accurately estimate the range of genetic distances corresponding to divergence events from taxa with different generation times, deme sizes, life expectancy, vagility and dispersal, body size, breeding strategies, and so on. It is well documented that there are different rates of molecular evolution in different taxa and understanding these differences will be valuable in fine-tuning the probability of genetic isolation (Steppan et al. 2004) in allopatric and parapatric phylogroups. Sequence data must involve only single speciation events (i.e., sister taxa comparisons; Table 3 and Fig. 1, to be discussed below). Data from sister species to serve as a standard will be most valuable if they represents distance values from different markers and loci such as mtDNA, introns, exons, amplified fragment length polymorphism (AFLP)-type data (Giannasi et al. 2001), and Y-linked markers (Cathey et al. 1998). Minimally, there should be ≥1 marker each for mtDNA and nuclear DNA. For allopatric populations, use of phylogenetic principles and methods to generate gene trees that document sister taxa and examples of paraphyly (Hoffmann and Baker 2003) can be used to resolve species boundaries. This is compatible with the Phylogenetic Species Concept where nonmonophyletic populations would be recognized as different species (Hoffmann and Baker 2003).
Genetic species, morphological analysis, and species-level names
If genetic data are interpreted as justifying specific recognition of a previously unrecognized species, then it becomes important to document the level of morphological distinction that is present in the 2 newly distinguished sister species (Wiens and Penkrot 2002). It is important to note that the magnitude of morphological distinction that distinguishes the 2 is probably small because these were recognized as conspecifics before genetic data documented species distinction. The most powerful way to determine morphological differences between the 2 species is to use museum voucher specimens for which genetic data document genetic species status. In many cases, there will be specimens in natural history museum collections for which there are no genetic data, and morphological features of individuals documented by genetic data will be valuable in identifying these voucher specimens to specific status.
When a previously unrecognized species is identified using genetic data, it is necessary to determine the appropriate species-level name. This procedure is outlined in the 4th edition of the International Code of Zoological Nomenclature (International Commission on Zoological Nomenclature 1999). In most cases, the previously unrecognized species will be a genetically defined phylogroup within a species recognized in Wilson and Reeder (2005). The holotype for that species will be a member of 1 of the 2 phylogroups and that phylogroup will retain that species name. Basically, if there is a species-level name or names (either junior synonyms or currently recognized subspecies) available that was described based on a specimen of the alternative phylogroup then the oldest available name has priority (senior synonym) and would be elevated as the appropriate species name. Species-level names that might be applicable are listed as synonyms (Wilson and Reeder 2005); however, it is necessary to determine if the synonyms present were based on a specimen of the newly recognized species (Gardner and Hayssen 2004). If no species-level name is available, then it is appropriate to describe and name a new species following the rules of the International Code of Zoological Nomenclature (International Commission on Zoological Nomenclature 1999). Description of a new species is complex and care should be taken to make sure that the rules to create a valid species-level name are followed.
Phylogroups and undescribed species
Members of the bat genus Carollia provide an example of the type of genetic variation present in mammalian species and the implication of that variation to the Genetic Species Concept and to current species catalogues (e.g., Wilson and Reeder 1993, 2005). Members of Carollia are among the most commonly collected species in the New World tropics, resulting in sufficient voucher specimens in museums to study intra- and interspecific geographic morphological variation (Fleming 1988; McLellan 1984; Pine 1972). It might be expected that the number of species and species boundaries would be well defined for such an often-studied common bat, but this is not the case. In 1993, only 4 species (brevicauda, castanea, perspicillata, and subrufa) were recognized (Koopman 1993). Today, 9 species are recognized, with the description of colombiana (Cuartas et al. 2001), sowelli (Baker et al. 2002), monohernandezi (Muñoz et al. 2004), manu (Pacheco et al. 2004), and benkeithi (Solari and Baker 2006). Three of these new species (colombiana, manu, and monohernandezi) were recognized by classical skin and skull methods; 2 of these species (sowelli and benkeithi) probably would not have been recognized by studies of museum voucher specimens without genetic data and were recognized through application of the Genetic Species Concept.
As often will be typical, the initial description of genetic variation in Carollia had small samples (n = 10) and limited geographic sampling (Wright et al. 1999). Even with that small sample and only data from the cytochrome-b gene, because of paraphyly in C. brevicauda it was possible to detect that C. brevicauda was probably comprised of 2 different species (ultimately C. brevicauda and C. sowelli). To make all C. brevicauda (sensu Koopman 1993) monophyletic, which is a standard for species definitions, would require the inclusion of C. perspicillata (Fig. 1). C. brevicauda and C. perspicillata are sympatric (often collected in the same mist net at the same time) over a wide geographic area including much of South America and Middle America. Based on morphology, these 2 species do not hybridize (Pine 1972); therefore it would be inappropriate to include C. brevicauda and C. perspicillata in a single species. Morphological features distinguish the clade of C. brevicauda sister to C. perspicillata from the clade basal to these 2 (Fig. 1), evidence that further justifies recognition of the basal clade as a separate species (Baker et al. 2002). No species-level name was available for members of the basal clade. Therefore, a new species, C. sowelli (Baker et al. 2002), was described. Wright et al. (1999) had suggested further study was merited based on the genetic distance between the 2 specimens of C. castanea. Intraspecific genetic distance (8.7%) between the 2 individuals of C. castanea was greater than the values that distinguished members of the only other sister-species comparisons in that tree (perspicillata-brevicauda, 4.1–4.8%).
A follow-up paper on genetic variation in Carollia (Hoffmann and Baker 2003; Fig. 1) was designed to better understand phylogenetic clades in 5 species of Carollia and examined 66 specimens of the genus to provide a more detailed geographic sampling. That study defined the species boundaries between C. sowelli, which was distributed from Panama north through Mexico, and C. brevicauda, which was distributed from Panama south into South America, and it showed that C. brevicauda and C. sowelli both were sympatric with C. perspicillata. It also documented ≥4 phylogroups within C. castanea with divergence values > 5% and that those phylogroups were more distant from each other than were C. sowelli, C. brevicauda, and C. perspicillata from each other. The type locality for C. castanea is in Central America, and no other species-level names are available for C. castanea (Pine 1972).
It is important to have both nuclear and mitochondrial markers to document presence or absence of species. For the phylogroup of C. castanea that was distributed in Peru and Bolivia, such a nuclear marker existed in the presence of a unique karyotype, different from that of the other members of the genus. Although there was minimal morphological divergence between this phylogroup with a unique karyotype and the phylogroup of which the holotype of C. castanea was a member, concordance between mitochondrial and nuclear markers was used as justification for description of the species C. benkeithi (Solari and Baker 2006). The description of C. benkeithi leaves 3 other phylogroups within C. castanea (C. castanea sensu stricto and clades “1?” and “2?” in Fig. 1) that have genetic distance values > 5% in the mitochondrial cytochrome-b gene. Interpretation of these high values remains problematic because there are few data from the nuclear genome or from morphology that distinguish clades “1?,” “2?,” and C. castanea sensu stricto from each other. In this case sensu stricto refers to the phylogroup that contains the holotype for the species-level name castanea. If it should prove that the level of genetic distance in the nuclear genome is typical of, or greater than, the amount that distinguishes other sister species of Carollia or other phyllostomid bats, then it will be appropriate to recognize these 3 phylogroups as species: C. castanea, which will then be distributed in middle America and northern South America; clade “1?” presently known only from Panama; and clade “2?” from eastern Ecuador and Peru (R. J. Baker, in litt.). Other genetic variation present in species of Carollia indicate 2 phylogroups in C. sowelli (labeled 1 and 2 in C. sowelli clade; divergence values = 3.6%), 2 in C. brevicauda (labeled 1 and 2 in C. brevicauda clade; 2.3%), and 3 less well-defined phylogroups in C. perspicillata (clades 1, 2, and 3; ≤2.4%; Fig. 1). Even though the probability of 2 species is less than if the distance value was > 5%, these phylogroups could be explored for limits of geographic distribution, concordance of nuclear and mitochondrial genetic markers, extent of hybridization, and sympatry at boundaries at geographic limits. Genetic data would then form the basis for decisions relative to the presence or absence of species, subspecies, or non–taxonomically recognized geographic variation. Such data from phylogroups with smaller distance values for the cytochrome-b gene would be valuable in understanding how often this magnitude of genetic divergence is accompanied by genetic isolation and speciation. This information is important in further documentation of the underestimation of the number of species present in the species listed in Wilson and Reeder (2005).
In Bradley and Baker (2001) and this paper, we have summarized data available for genetic distance values of the cytochrome-b gene for sister species (Table 3). Use of sister species helps filter out the amount of genetic change that is phyletic rather than that involved in evolution of genetic isolation and speciation. In Fig. 1, the only example of sister-species relationships where morphologically determined species were present for examination are C. perspicillata-brevicauda. We have marked the portion of the tree during which all members of C. brevicauda (A in Fig. 1) and all members of C. perspicillata (B in Fig. 1) examined in the gene tree could have accumulated genetic change that produced genetic and reproductive isolation for C. brevicauda and C. perspicillata from each other. The distance values representing the period when genetic isolation evolved between C. brevicauda and C. perspicillata are relatively short in comparison to their intraspecific values (Fig. 1). The point of this observation is that if the distance values in this gene tree have predictive value about the time required for speciation, that time is small relative to the overall changes in the remainder of the tree revealing genetic diversification in Carollia. Avise and Walker (1998; Avise 2004:352) calculated a temporal ceiling and floor (our A and B branches in Fig. 1) on the duration of speciation. This calculation was derived from an estimate of separation between sister species minus the estimate for intraspecific variation. We agree that this is the most accurate way to calculate the duration of speciation. We considered duration of speciation for sister species as an alternative to the 1st step value of genetic distance between sister species for application of the Genetic Species Concept. Calculation of this value for the sister species C. brevicauda and C. perspicillata would reduce the distance values substantially. However, the ceiling–floor values are dependant upon sample size, and representation of the intraspecific diversity will vary accordingly. We think the most pragmatic 1st step is to use the >5% genetic distance for the cytochrome-b gene because in many cases there will not be an adequate estimation of intraspecific variation, plus the >5% is an acknowledged crude estimate and is not an end point. Calculation of the duration of speciation values will be an estimation of time associated with accumulation of genetic changes required for isolating mechanisms to evolve, as well as an estimate of a paleontological time frame during which the genetic changes of speciation occurred (Klicka and Zink 1997).
Consequences of Implementation of the Genetic Species Concept
Number of recognized species
In a comparison between the Biological Species Concept and the Genetic Species Concept, the beginning and end points of speciation will be congruent in definition of species. Panmictic populations with unlimited gene flow will be recognized as conspecifics, and populations that are totally isolated (genetic and reproductive) will be recognized as 2 distinct species. Many phylogroups present in mammals and morphologically distinct taxa (Van Gelder 1977) will not be totally reproductively isolated but will be sufficiently genetically isolated to provide protection of integrity of alternative gene pools. The extent that this is true will be the extent that the strict application of the Biological Species Concept (sensu Mayr 1942) and the Genetic Species Concept will produce different conclusions on the number of species present and the geographic limits of species. Application of the Genetic Species Concept will result in a greater number of recognized species than will the Biological Species Concept.
Based on our discussion of the BDM model, implementation of the Genetic Species Concept has resulted in a substantial number of unrecognized cryptic species (Fig. 1). Nonetheless, determining specific status of allopatric populations using the Genetic Species Concept will be similar to application of the Morphological Species Concept. The systematic community today accepts that if a certain level of morphological divergence distinguishes 2 allopatric populations, then 2 biological species exist. Although that conclusion creates stability and, from a probability standpoint, may or may not be correct >50% of the time, in each case justification involves few or no supporting data that document reproductive isolation (see “Application of the Genetic Species Concept” for our method of discerning species presence–absence using genetic distance values typical of sister species). In our comparison of the Genetic Species Concept and the Phylogenetic Species Concept (Table 1) the greatest number of species probably will be recognized by the Phylogenetic Species Concept because, at least in some definitions, application of that concept is not concerned with evidence of reproductive or genetic isolation. This comparison is not simple, however, because the definition of a species in the Phylogenetic Species Concept is so variable (Wheeler and Meier 2000).
Implications of the Genetic Species Concept to higher zoological taxa
If the hypothesis of Wilson et al. (1975) and Fitzpatrick (2004) that mammals evolve hybrid sterility faster than do other vertebrates through genetic divergence as proposed in the BDM model is true, then there may be more rapid speciation in mammals than in other vertebrate groups. Such a difference would be compatible with Patterson's (2000) observation concerning the rate of descriptions of new species of mammals versus birds. Nonetheless, birds, amphibians, reptiles, and fishes will likely have a pattern of underestimation adjusted for the rate of evolution of hybrid sterility and events that cause allopatric populations, relative to that observed for mammals (Avise and Walker 1999). For invertebrate taxa, because they have been studied less intensively than mammals and because generation time may be much shorter resulting in a faster rate of genetic evolution, which can result in genetic isolation, we hypothesize that the underestimation of biodiversity is significantly greater than for mammals.
Level of research efforts required to implement the Genetic Species Concept
The level of genetic study that we propose for each species of mammal (of which there are >5,000—Wilson and Reeder 2005) will require a substantial amount of labor and resources. But that is based on our current methods and, even with those, almost every issue in the Journal of Mammalogy, Molecular Ecology, and Molecular Phylogenetics and Evolution, for example, contains multiple publications using genetics to define species boundaries and species. DNA sequencing of whole genomes for multiple species is becoming less expensive, easier, and faster (Chan 2005; Zwick 2005), and such technological advances will result in a new research landscape for the interface of genetic information and studies on mammalian systematics, biodiversity, speciation, and so on. As a result, genetic profiles will soon be generated for many, if not most, mammalian species. A primary limiting factor will be availability of tissues for DNA isolation from a sufficient number of specimens and geographic localities to document the level of genetic isolation, hybridization, introgression, species boundaries, and so on for mammals. Technical advances are needed to be able to perform these studies on museum voucher specimens, and such advances will likely soon occur. Although use of sequence data from museum voucher specimens would not entirely solve the need for additional collecting, it would address the majority of issues concerning species diversity.
An aspect of this change in the study of mammalian diversity is that many new “mammalian systematists” will have little knowledge of the natural history and diagnostic features of the organisms they are studying (Schmidly 2005). This new concept of an organism may be a bit of tissue in the bottom of a plastic cryotube. It is our opinion that mammalian systematics must retain its historical association with and dependency on museum voucher specimens. We hope that this new breed of systematists will embrace the museum concept and cooperate with a museum accredited by the American Society of Mammalogists or other appropriate society to document their studies by depositing voucher specimens and tissues (Ruedas et al. 2000; Salazar-Bravo et al. 2006). This action not only will serve to validate their current studies but will provide material for future studies.
Acknowledgments
We thank S. Hoofer, D. Wilson, J. L. Patton, J. Coyne, E. Lessa, P. Larsen, F. Hoffmann, H. Genoways, C. Phillips, R. Chesser, J. Bull, J. Bickham, S. Solari, L. Ammerman, R. Pfau, A. Brown, F. Bronson, M. Knapp, H. Mantilla, H. Meeks, V. Swier, R. K. Baker, and the Principles of Systematics class at San Angelo State University for discussions and critical reviews. We thank F. Hoffmann for assistance with Fig. 1. We thank L. Bradley and T. Ginter for technical assistance. Financial support was provided by J. Sowell, A. Brown, the Texas Tech University's Biological Database Program, and the National Institutes of Health (DHHS A141435-01) to RDB.
Literature Cited
Fig. 1.—Phylogram of genetic relationships among species of the bat genus Carollia based on complete DNA sequences from the mitochondrial cytochrome-b gene. This maximum-likelihood tree depicts phylogroups and genetic distance (horizontal length of each clade) pertinent to illustrating the application of the Genetic Species Concept. This figure was developed from data and figure 3 of Hoffman and Baker (2003). Information from a parsimony analysis (significant bootstrap values ≥ 70 above a clade) and a Bayesian analysis (significant posterior probability values ≥ 95 below a clade) were superimposed on the tree to illustrate support values for nodes and clades. Values not provided were not statistically significant. Combined distance values of A (shared common ancestry of all specimens of C. brevicauda) and B (shared common ancestry of C. perspicillata) represent the “duration of speciation” value of Avise and Walker (1998). Abbreviation for each specimen: Cbr = C. brevicauda, Cp = C. perspicillata, Cso = C. sowelli, Csu = C. subrufa, Cca = C. castanea, and Cbk = C. benkeithi. Numbers by vertical lines within species identify intraspecific phylogroups referred to in the text. Glyphonycteris sylvestris was used as the outgroup

Table 1.—Criteria for recognition and comparisons between the Biological, Morphological, Phylogenetic, and Genetic Species Concepts (modified from Cracraft 1997:333). The Genetic Species Concept is as defined in this paper. Criteria 1–9 are from Cracraft's original table, except that 4 has been restructured. The interpretations for 1–9 of the Biological Species Concept and the Phylogenetic Species Concept are from Cracraft's table, whereas the Morphological Species Concept interpretations are by the authors of the present study. All interpretations for 10–14 and the Genetic Species Concept in 1–14 are those of the present authors

Table 2.—Characteristics of hybridization documenting absence of reproductive isolation in 4 species and 2 possible species of mammals. Note how characteristics of hybridization vary among species. Hybridization in mammals is common even in distantly related taxa (Van Gelder 1977)

Table 3.—Genetic variation at different taxonomic levels from the mitochondrial cytochrome-b gene for marsupials, bats, rodents, and artiodactyls. Values were obtained from published studies; in some cases, we calculated means and ranges. Values are sample size in parentheses, followed by mean and range of DNA sequence values as a percentage

Table 4.—Species of mammals for which data on genetic variation (%) in cytochrome-b data has been generated from different geographic localities that could potentially identify phylogroups that represent different species. n = number of specimens sampled. Potential unrecognized species are the number of phylogroups in that species with variation > 5% minus 1 for the phylogroup that contains the holotype for the species