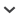Landscape genetics is a rapidly evolving interdisciplinary field that integrates approaches from population genetics and landscape ecology. In the context of habitat fragmentation, the current focus of landscape genetics is on assessing the degree to which landscapes facilitate the movement of organisms (landscape connectivity) by relating gene-flow patterns to landscape structure. Neutral genetic variation among individuals or direct estimates of current gene flow are statistically related to landscape characteristics such as the presence of hypothesized barriers or the least-cost distance for an organism to move from one habitat patch to another, given the nature of the intervening matrix or habitat types. In the context of global change, a major challenge for landscape genetics is to address the spread of adaptive variation across landscapes. Genome scans combined with genetic sample collection along environmental gradients or in different habitat types attempt to identify molecular markers that are statistically related to specific environmental conditions, indicating adaptive genetic variation. The landscape genetics of adaptive variation may also help answer fundamental questions about the collective evolution of populations.
Interdisciplinarity lies at the heart of landscape genetics, a field described as an “amalgamation of molecular population genetics and landscape ecology” (Manel et al. 2003). Storfer and colleagues (2007) proposed a more distinct definition of landscape genetics, stating that the field comprises “research that explicitly quantifies the effects of landscape composition, configuration and matrix quality on gene flow and spatial genetic variation.” In a broader sense, landscape genetics builds from those studies that combine population genetic data, adaptive or neutral, with data on landscape structure (Holderegger and Wagner 2006). The matrix in the quotation above defines the often-hostile space that separates the patches of a species' habitat in a given landscape (figure 1; Turner et al. 2001).
Figure 1.
Two commonly used landscape representations and their implications for assessing structural landscape connectivity and landscape change. The patch-matrix representation (top left) considers only patches of suitable habitat (a–d) as islands in an ocean of non -habitat (matrix). The movement of organisms is expected to depend on the physical distance between patches. Landscape change is limited to habitat gain or loss through expansion or shrinkage (c), appearance (e) or attrition (b), and subdivision (d) of individual patches (top right). In the mosaic representation (bottom left), each patch belongs to one of several habitat types. Although patches a and d are at about the same distance from patch c, the road between patches c and d may act as a barrier to many organisms; the movement of organisms is expected to depend on matrix resistance or on the nature of the intervening habitat types. Landscape change in the mosaic representation consists not only of gains and losses of individual habitat types but also of different types of transitions from one type to another (bottom right).

The incorporation of the matrix into landscape genetics is a discriminating difference between landscape genetics and population genetics. At most, the latter includes the stretches of land between occupied habitat patches as a simple function of geographical distance; in contrast, in landscape genetics the matrix is seen as a major determinant of biological and ecological processes at the landscape level, and the different quantities and qualities of the areas that separate habitat patches are quite important. For instance, a strip of woodland might not hinder the movement of a ground-breeding bird found in open grasslands, but it could severely limit the migration of meadow butterflies or even form a complete barrier to the dispersal of meadow-plant seeds by wind. Landscape genetics does not possess its own conceptual methodological framework or its own analytical or statistical tool kit, but combines approaches and methods from landscape ecology, population genetics, and spatial statistics. We argue that landscape genetics is not a scientific discipline in itself but rather provides a perspective for examining spatiotemporal processes such as habitat fragmentation (Fahrig 2003). The spatial scale and extent at which landscape genetic research occurs are predefined by the species-specific biological and ecological process under study, and by the spatial dimension at which operational practical measures can be taken. The “landscape” of landscape genetics therefore often consists of catchments, one or several valleys, hundreds of square kilometers of forest area, a part of a motorway and its hinterland, or an area of urban sprawl around a city center.
A current question of landscape genetics: Inferring and testing landscape connectivity
Landscape ecology and population genetics naturally converge in the exploration of how habitat loss and the spatial isolation or fragmentation of habitats affect the movement of species across landscapes. The constraints that landscape patterns and the matrix impose on dispersal—and thus on the distribution of animals, plants, and their genes in a landscape—have implications for the dynamics and persistence of populations as well as for local species diversity (Fischer and Lindenmayer 2007). As a consequence, the maintenance of the exchange of individuals or genes among populations in different habitat patches and the restoration of such exchange (i.e., defragmentation) are highly relevant in conservation management. Landscape connectivity has been defined as the interaction between the movement behavior of organisms and the structure of the landscape (Merriam 1984, Goodwin 2003). Landscape connectivity thus has a structural and a functional component (Brooks 2003), and landscape genetics is ideally suited for testing the effect of structural landscape connectivity (e.g., the distance between habitat patches and the nature of the intervening habitat types) on functional landscape connectivity (i.e., dispersal and gene flow between habitat patches). The important question, however, is this: how can we reliably assess and predict functional landscape connectivity?
Landscape ecology and population genetics address this question from different perspectives. Landscape ecology has developed a suite of tools (i.e., landscape metrics; for reviews, see McGarigal 2002, Li and Wu 2004, implemented, e.g., in the FRAGSTATS software [McGarigal et al. 2002]) for quantifying landscape patterns, and thus for measuring structural landscape connectivity. Landscape metrics are commonly calculated from habitat maps, either in the form of habitat patches floating in a matrix of unsuitable habitat or in the form of a mosaic of different habitat types, such as land-use and land-cover maps. Figure 1 illustrates the difference between these landscape representations, both for landscape composition and landscape change. The patch-matrix representation considers only the target habitat type, which may undergo habitat loss, gain, or fragmentation over time (figure 1, top; Fahrig 2003). Connectivity can be assumed to depend on interpatch distance only. All of the surrounding matrix is assumed to be equally inhospitable, so the habitat patches resemble islands in an ocean. The mosaic landscape model considers all habitat types and distinguishes between different types of transitions over time (figure 1, bottom). In this model, connectivity depends not only on the distance between suitable habitat patches but also on the nature of the intervening habitat types. Both landscape representations assume that habitat patches are internally homogeneous and that there are crisp boundaries at the transition between habitat patches and matrix, or between different types of land use or land cover.
Population genetics, on the other hand, studies fine-scale genetic structure (i.e., how genetic variation is distributed in space) and current gene flow in various ways. As gene flow comprises the dispersal of organisms (including seeds in plants) or the movement of genes alone (pollen in plants and haploid propagules in cryptogams), it provides a direct measurement of functional connectivity (Holderegger et al. 2007). In its full meaning, however, landscape connectivity refers to the interaction between structural and functional connectivity. Landscape genetics thus combines approaches from both population genetics and landscape ecology to address landscape connectivity (Allendorf and Luikart 2007).
Landscape genetics uses two approaches to study gene flow among populations. The first approach is an individual-based assessment of fine-scale genetic structure; the second addresses recent or current gene flow directly (figure 2).
Figure 2.
Currently used landscape genetic approaches to infer barriers to gene flow. In the three figures, the black line refers to a specific landscape feature, such as a river; the filled circles refer to the adult individuals of a study species; and different gray shadings of these circles refer to genetically similar genotypes. (a) Approach based on individual genetic distances. Here, the genetic distances among all sampled individuals are determined and correlated with landscape structure. In the present case, the largest genetic distances occur across the landscape feature, that is, the hypothesized barrier (hatched double-headed arrow). (b) Recent gene flow assessed by assignment tests. The individuals are grouped into two predescribed populations on either side of the landscape feature. An assignment test identifies one individual (circle with solid outer line) as a recent immigrant from the other side of the landscape feature (arrow). (c) Current gene flow assessed by parentage analysis of offspring (squares). The hatched offspring has one parent on the other side of the landscape feature (arrow). The latter two methods indicate the occurrence of recent and current gene flow across the landscape feature, respectively.

The individual-based approach (figure 2a) analyzes relationships between genetic distances and cost distances among individuals. It has so far been applied mainly to animals, where many individuals of the study species are sampled within the study landscape. This does not necessarily mean that animals have to be captured; one can also use noninvasive methods such as sampling feces or trapping hairs of mammals to obtain material for DNA (deoxyribonucleic acid) extraction. In the next step, the genotypes of the sampled individuals are determined with highly variable molecular markers (box 1), and a metric of genetic distance is calculated among all possible pairs of individuals in the sample set, resulting in a matrix of pairwise genetic distances. In parallel, the landscape structure is analyzed. In the simplest way, multiple landscape variables (e.g., the amount of grassland, hedgerows, woodland, or settlements between genetic sampling sites; road density; topography; moisture gradients) are quantified in a geographic information system (GIS).
Box 1. The current molecular genetic toolbox of landscape genetics.
Because landscape genetics considers relatively small spatial scales, individuals of a study species are mostly closely related, so that the genetic differences among them are small. Molecular genetic analysis hence relies on highly variable molecular markers that provide enough power of resolution. Currently, landscape genetics typically uses two different genetic marker types, namely, single sequence repeats (SSRs) and amplified fragment length polymorphisms (AFLPs), which fulfill this requirement (for detailed descriptions see Lowe et al. [2004] and Allendorf and Luikart [2007]).
Microsatellites or SSRs are codominant, selectively neutral markers, located on the nuclear DNA and showing Mendelian inheritance. In codominant markers, both alleles at a heterozygote locus are identified, and heterozygote individuals can therefore be determined. Microsatellites consist of DNA stretches of several to many repeats of two to six nucleotides. For instance, a heterozygote individual may show one allele with nine CA repeats (CACACACACACACACACA), while the second allele has 10 repeats (CACACACACACACACA CACA). The two alleles thus differ in their length, which can be assessed by gel electrophoresis or by using automated sequencing machines after multiplication in a polymerase chain reaction (PCR). Molecular primers to amplify microsatellites have to be specifically developed for each study species de novo, because they are transferable only among closely related species. Between 6 and 15 SSRs are typically used in a landscape genetic study.
Amplified fragment length polymorphisms provide an alternative molecular marker type, often used in plants. Hundreds of fragments, spread over the entire genome, are amplified in a complicated PCR approach. The amplified fragments are anonymous—that is, their locations on the genome are unknown. Commonly, AFLPs are assumed to be selectively neutral, dominant markers. In dominant markers, it is not possible to discriminate heterozygotes. Basically, the raw data of an AFLP study look like vertical barcodes; the barcode lines represent DNA fragments of different lengths. The barcodes of individuals are different, and the simple fragment presences and absences are scored per individual in a 0/1-matrix. AFLPs are detected by fragment length analysis in gel electrophoresis or on automated sequencing machines.
Codominant single nucleotide polymorphisms (SNPs) provide a new type of molecular markers hardly used in landscape genetics to date. Individuals of the same species share many DNA sequences that are almost identical and differ only at a few positions within the sequences. At these sites, the two copies of a gene in a heterozygote individual show different nucleotides, whereas a homozygote individual shows only a single nucleotide. Finding SNPs first requires the sequencing of many genes of a genome, which is a cost- and time-intensive task. As more DNA sequences of nonmodel organisms are deposited in open-access DNA databases, the use of SNPs in landscape genetics may increase rapidly. SNP detection is fast when using specific equipment (e.g., real-time PCR, pyrosequencing).
In contrast to the above three biparentally inherited marker types, the mitochondrial DNA (mtDNA) of animals and plants and the chloroplast DNA (cpDNA) of plants are uniparentally inherited (usually from the mother), and they are nonrecombinant. This means that they are transmitted unchanged from the mother to her offspring, which, in principle, makes them useful for the detection of dispersal events. Unfortunately, mtDNA and cpDNA often do not provide enough genetic variation among individuals at the spatial scales of landscape genetic studies. However, if there is enough mtDNA or cpDNA variation among the individuals within a landscape, dispersal can be readily determined. Variation in mtDNA and cpDNA is assessed by DNA sequencing or fragment analysis using restriction enzymes, but uniparentally inherited SSRs and SNPs can be used as well.
In a variation of this approach, least-cost paths are determined (Adriaensen et al. 2003). In principle, different species-specific resistance weights (which quantify how permeable an element is for a particular species) are given to particular landscape features, land-use, or land-cover types between sampling points. In this way, the most probable migration route is identified. For instance, Cushman and colleagues (2006) used GIS to derive a set of 110 resistance surfaces representing alternative hypotheses about the relative importance of elevation, slope, roads, and land-cover types on black-bear (Ursus americanus) movement. Causal modeling based on Mantel tests indicated that land cover and elevation most influence the movement of black bears, whereas models that included isolation by hypothesized barriers or by geographical distance were not supported.
In both types of landscape assessments, as illustrated in the black-bear example, pairwise genetic distances are finally statistically correlated with landscape variables, commonly by partial Mantel tests (i.e., multiple matrix correlations; Legendre et al. 2002). Partial Mantel tests identify the landscape variables that explain significant levels of the genetic distance among individuals. The pure geographical distance between sampling points is often used as a null model in these analyses, because population genetic theory predicts that genetic distances among individuals will increase with increasing geographical distance (Allendorf and Luikart 2007). Although the use of partial Mantel tests is controversial (Castellano and Balletto 2002), studies that used partial Mantel tests produced meaningful results. For example, Coulon and colleagues (2004) demonstrated that least-cost paths, as determined by the straight-line distance through suitable habitat between sampling points, explained the genetic distances among roe deer (Capreolus capreolus) individuals better than did geographical distances alone.
A problem with this approach is the a priori selection of those landscape features that should be included in the analyses, as the relevant landscape features are often not known in advance. Cushman and colleagues (2006) partially circumvented this problem by considering a wide range of possible landscape features. Moreover, the direct comparison of alternative hypotheses about the effect of different landscape characteristics on black-bear movement showed that, while most hypotheses proved statistically significant, candidate models differed strongly in their empirical support. Statistical significance alone is not a measure of ecological relevance, and landscape genetic studies could profit greatly from applying model-selection procedures (e.g., adopting the framework of the information theoretic approach by Burnham and Anderson [1998]).
Alternatively, even simple studies on fine-scale genetic structure may provide valuable insight into biological and ecological processes in a landscape, if the boundaries of objectively inferred genetic groups coincide with supposed barriers in the landscape such as roads or settlements. In the roe-deer example, Coulon and colleagues (2006) used Bayesian clustering approaches (e.g., STRUCTURE software, Pritchard et al. 2000; GENELAND software, Guillot et al. 2005) to show that the boundaries of genetic groups within the study landscape followed the course of a river and a motorway. These landscape features were hence identified as barriers to roe-deer movement. Landscape genetics uses a multitude of similarly sophisticated statistical methods (enumerated in Manel et al. [2003], Storfer et al. [2007]).
Any individual-based assessment of fine-scale genetic structure is correlative in nature and infers gene flow indirectly from genetic distances among individuals. In contrast, the second approach in landscape genetics assesses current or recent gene flow patterns directly. Assignment tests, which infer first-generation migrants (Rannala and Mountain 1997, Piry et al. 2004 [GENECLASS software]), are often used to assess dispersal in landscape genetic studies on animals and plants (figure 2b; Manel et al. 2005). In this approach, many individuals (usually about 30) are sampled from every population of a study species within a landscape. Using highly variable mo lecular markers (box 1), the genotypes of all sampled individuals are then assessed. From the allele frequencies per population, and by applying maximum-likelihood statistical methods, it can be determined which individual within a given population possesses a genotype that better matches the genotypes of another population. In essence, “home” and “away” genotypes are identified. An “away” genotype is identified as a migrant from one known population to another known population. This method refers to recent migration because (a) the migration event has already happened and was not directly observed, and (b) the footprint of migration will be rapidly erased in only one or few generations owing to the immigrant's mating with individuals from the local population. Using such an assignment test, Kraaijeveld-Smit and colleagues (2005) found almost no recent individual movement among populations from several river catchments in the Mallorcan midwife toad (Alytes muletensis), showing that the landscape between rivers formed a complete barrier to recent gene flow.
An alternative approach for studying current gene flow, parentage analysis (figure 2c; Sork et al. 1999), has been used mainly in plants (paternity analysis to detect gene flow by pollen and maternity analysis to detect dispersal by seed). For paternity analysis, open-pollinated seeds (i.e., the offspring) are sampled from several mother plants, and plant tissue from all adult individuals within the study area is collected (i.e., all potential fathers or pollen donors). The genotypes of all sampled offspring, mothers, and potential fathers are then assessed (box 1), and the most probable father is determined by exclusion using maximum-likelihood methods (e.g., CERVUS software; Marshall et al. 1998). Paternity analysis thus provides trajectories of current gene flow by pollen, and it identifies the amount of gene flow from outside the study area (i.e., pollen immigration) when no suitable father genotype can be identified within the sampled area. In a population of the wild service tree (Sorbus torminalis) that was part of a larger regional metapopulation, Hoebee and colleagues (2007) found more than 30% of gene flow by pollen from outside the sampled area, whereas pollen immigration dropped to about 5% in a spatially separated small population. This result clearly indicates the negative effect of spatial isolation on gene flow among populations. In maternity analysis, dispersed seeds are trapped all over the study area, and the most likely mother is identified in the same way as in paternity analysis (using maternal tissue from the seed coat). Using maternity analysis, Godoy and Jordano (2001) showed that about 20% of the seeds trapped within a population of the shrub St. Lucie Cherry (Prunus mahaleb) originated from outside the population, indicating an unexpectedly high rate of seed immigration.
The two methods for direct estimation of gene flow have rarely been coupled with a detailed assessment of landscape structure in a statistical way. One reason for this shortcoming is that parentage analysis requires the sampling of all adults (i.e., parents), which obviously limits the manageable spatial extent of study areas and almost prevents the method's application to animals. The assessment of recent gene flow with assignment tests, on the other hand, relies on the assumption that the species is structured into discrete populations. When applied to species with a gradient-like population structure, such methods may produce spurious results (Cushman et al. 2006). For parentage analyses and assignment tests, a large number of migrants need to be found to disentangle the effects of different landscape features on gene flow. However, similar landscape analyses as made in studies based on individual genetic distances (see above) can, in principle, be used for the comparison of direct gene flow estimates with landscape structure. For instance, one could assess the correlation of inferred gene flow trajectories with the spatial expansion of landscapes features (such as postulated physical or behavioral barriers) and compare the result with a neutral yet spatially explicit model that assumes complete random gene flow among populations. In contrast to the individual-based approach, there is not yet a general consensus on a standard landscape genetic approach to evaluate data on current or recent gene flow.
In conclusion, landscape genetics has great potential to infer functional connectivity at spatial scales and for species for which ecological techniques are not applicable (e.g., radio tracking, global positioning system technology, and mark- recapture in animals [Cushman 2006]; single-source dispersal and pollen-dye experiments in plants [Nathan 2006]). For instance, landscape genetic approaches may be used to evaluate the success rate of connectivity or defragmentation measures already in place (figure 3) to quantify the spread of pest species (Storfer et al. 2007) or to monitor the spread of genes escaped from genetically modified species (Watrud et al. 2004, Reichmann et al. 2006). Many corresponding landscape genetic studies have been undertaken, and important results will soon be available.
Figure 3.
The assessment of the success rate of connectivity measures, such as wildlife bridges or overpasses over motorways, is a typical practical application of landscape genetic research. Photograph: Manuela DiGiulio.

One problem with landscape genetics is the need for replication of the study unit, that is, the landscape itself. If a fragmented landscape is compared with a highly connected landscape, the level of replication is actually a sample size of N = 1 per treatment (i.e., fragmented vs. connected). However, increasing replication often exceeds the workload that a single laboratory can manage.
A possible future question of landscape genetics: Adaptation to land-use and climate change
Landscape genetics uses neutral markers for assessing gene flow and functional landscape connectivity. By definition, neutral genetic variation is not subject to selection. Once an allele has been added to a local gene pool by immigration, it will remain in the gene pool of the population unless it is eliminated by stochastic processes (i.e., genetic drift). Natural selection, on the other hand, may lead to the elimination of less favorable immigrant alleles at adaptive loci within a few generations, thus eliminating the very traces of gene flow.
Additionally, gene flow is a whole-genome process, whereas natural selection is a specific process that acts on single genes or groups of genes involved in a particular physiological or morphological trait. By expanding from the investigation of neutral genes to the study of adaptive genetic variation, a new research field may open up for landscape genetics.
Land-use patterns are changing rapidly. Methods for studying gene flow can provide information on whether organisms keep track (by dispersal and migration) with the changing distribution and spatial arrangement of suitable habitat patches (Higgins et al. 2003). It is a different question, however, whether they could also keep track with these changes in an evolutionary way—that is, whether adaptation to a changing environment is or will be possible. A focus on adaptive genetic variation becomes paramount at larger spatial scales when considering climate change, which has important consequences for landscapes, organisms, and biodiversity (Walther et al. 2002). If the mean annual temperature at any location increases by 1.1 to 6.4 degrees Celsius within the 21st century (Solomon et al. 2007), previously well-adapted genotypes may lose their competitive advantage, and selection may favor other genotypes already present in the population or newly immigrating genetic variants (Thuiller 2007).
In Darwinian terms, genetic diversity should allow a species to adapt to new environments and thus maintain its evolutionary potential. However, the neutral genetic markers currently studied in landscape genetics do not allow researchers to infer adaptive genetic variation within populations or species, or their adaptive or evolutionary potential (Holderegger et al. 2006). Landscape genetics therefore will have to study different types of genetic markers—that is, markers relevant for adaptation and under natural selection—if it aims to address the question of adaptation in a spatially explicit landscape context.
From a landscape ecological perspective, the observed climate change requires us to consider gradual changes and heterogeneity both in space and in time. At the level of a single habitat patch, site conditions can no longer be assumed constant. At a regional scale, the area characterized by a specific set of climatic conditions generally tends to shift toward higher altitudes or latitudes (Bakkenes et al. 2002). The landscape ecology of climate change has important implications for population genetics. To be meaningful at the population level, climate-change scenarios need to be scaled down to the landscape scale and to biologically relevant factors (Prentice et al. 1992, Bakkenes et al. 2002). For instance, the length of the growing season (degree days) or the ratio of actual to potential evapotranspiration may be much better than mean annual temperature and precipitation for predicting the occurrence of many plant species. In addition, fluctuation from year to year may be more important than averages (Benedetti-Cecchi 2003). The magnitude of the expected changes in temperature and precipitation is not necessarily constant over larger areas but may vary considerably at a landscape scale as a result of topography, vegetation, and the presence of large water bodies. Jolly and colleagues (2005) showed that for the European Alps, the effect of the 2003 heat wave on vegetation growth, as estimated from satellite-derived photosynthetic activity, varied strongly with elevation, with changes in the effective growing season ranging between +64% for nival areas and −9% for colline areas. In particular, it may be the rare events, such as exceptionally dry summers, that will determine the composition of future species assemblages or ecosystems. The question is whether species will be able to adapt to changing environments. Indeed, rapid evolutionary change (Stockwell et al. 2003) due to exceptional drought has been shown to occur within only a few years in a North American annual plant species (Franks et al. 2007).
How should landscape genetics address the issue of adaptive variation in response to land-use change and gradual climate change? Genetic variation and differentiation of populations at adaptive traits have traditionally been assessed by quantitative genetic methods in common-garden experiments. These methods are notoriously labor and time intensive, and do not allow reference to the underlying genes per se (McKay and Latta 2002). Thus, these methods do not identify traceable molecular markers for adaptive genes, whose distribution and spread we could follow within and among landscapes. Modern genomic methods such as analysis of quantitative trait loci (i.e., genomic regions involved in the expression of a particular trait analyzed from known crosses), gene expression profiling (i.e., genes expressed under certain environmental conditions, as inferred from messenger RNA analysis), and candidate genes (known from the genome of model organisms such as Arabidopsis thaliana in plants) principally provide such molecular markers (for detailed descriptions, see Jackson et al. [2002], Vasemägi and Primmer [2005], Kohn et al. [2006], and Bouck and Vision [2007]). However, a major limitation of these genomic methods is that it is difficult to associate them with environmental variation in natural landscapes and to apply them to multiple populations, as will be necessary in landscape genetics.
An alternative approach using genome scans and genetic sampling along ecological gradients or in different habitat types promises to change this picture soon (Vasemägi and Primmer 2006, Reusch and Wood 2007). This landscape-based approach makes use of a massive screening of molecular markers in the genome to infer genes or loci that show signs of adaptive selection, and to correlate them with data on environmental heterogeneity. Hence, a large number of individuals and populations are sampled from different habitat types or along environmental gradients within replicated landscapes (figure 4). The corresponding environmental variation can be characterized in GIS. All samples are screened with principally neutral genetic markers (e.g., tens of microsatellites, hundreds of AFLPs or SNPs; box 1) over the whole genome (i.e., a genome scan; Storz 2005). The data set is then screened for particular loci that show a higher genetic differentiation among populations or habitat types as compared with the vast majority of neutral markers by using sophisticated statistical approaches (e.g., software FDIST, Beaumont and Balding 2004). The deviating differentiation of these “outlier loci” as compared with a model of neutrality (Storz 2005) is indicative of natural selection acting on them or of their genetic linkage (i.e., physical proximity within the genome) with genes under selection. Statistical correlation of outlier loci with environmental data from the locations where the samples had been taken then reveals which local environmental conditions or landscape characteristics are related to which outlier loci. We could thus identify and subsequently genetically characterize loci indicative of adaptive genetic variation related to certain environmental features within landscapes much faster than the above-mentioned genomic methods allow. It is an important advantage of this approach that it provides clear clues to those environmental factors that act as selective forces.
Figure 4.
A brief conceptual description of the use of genome scans to identify genetic markers that show signs of adaptive selection. (a) In a first step, samples (small filled circles) are taken from populations in different habitat types (indicated by differently shaded, larger circles) or spread along an environmental gradient and repeated over several landscapes. (b) In a second step, many principally neutral genetic markers, such as amplified fragment length polymorphisms, are determined for each individual sample. “Outlier loci” (arrows) indicative of natural selection—that is, loci showing a higher genetic differentiation among populations or habitat types than expected under a model of neutrality—are statistically identified (the dotted grey line indicates the statistical confidence limit). These outlier loci are either adaptive themselves, or they are linked to adaptive genes in the genome. (c) In the third step, DNA sequences of outlier loci are characterized, and easily applicable molecular markers are developed for further investigation.

So far, only a handful of studies have used this approach in non–model organisms and in real landscapes. Using a genome scan, Bonin and colleagues (2006, 2007) identified several AFLP markers that were linked to altitude in the common frog (Rana temporaria). Although there is yet to be final proof that these loci really affect the fitness of individuals or populations, studies like the one on the common frog could enable us to study gene flow not only at neutral markers but also at adaptive markers, because the generated molecular markers for genes involved in adaptation would be easily traceable across landscapes. It will then become possible, first, to investigate the relationship between adaptive variation and environmental gradients or different habitat types and, second, to address whether gene flow spreads genes relevant to adaptation over a landscape under a climate change scenario. The latter is arguably one of the most exciting topics in today's evolutionary biology and ecology (Reusch and Wood 2007).
An answer to an old question: Landscape genetics' contribution to basic evolutionary science
As discussed above, novel approaches linking spatially explicit environmental analysis with molecular genetics could offer effective means to study the spread of adaptive genes across landscapes. As a contribution to basic evolutionary science, the landscape genetics of adaptive variation may provide a much-needed empirical basis for answering the fundamental evolutionary question of the collective evolution of populations (Rieseberg and Bourke 2001). Gene flow and the spread of advantageous mutations across populations and landscapes is theoretically an important cohesive force in evolution. Without such a role for gene flow, one could argue that populations or groups of populations (e.g., metapopulations) form separate, individually evolving gene pools. Although the question of the collective evolution of populations (i.e., populations of a species evolve in the same direction) is central to Darwinian evolutionary theory, we know almost nothing about the pace of the spread of adaptive genes or mutations across the populations of a species. Rieseberg and Bourke (2001) recently emphasized this point: “We also note that the traditional role of gene flow as a force that constrains differentiation due to genetic drift or local adaptation has been over-emphasized relative to its creative role as a mechanisms for the spread of advantageous mutations.”
Acknowledgments
We thank all our colleagues working in landscape genetics for stimulating discussions and continuous intellectual support, as well as Felix Gugerli and three anonymous referees for valuable comments on an earlier version of the manuscript.