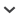Eastern North America receives elevated atmospheric mercury deposition from a combination of local, regional, and global sources. Anthropogenic emissions originate largely from electric utilities, incinerators, and industrial processes. The mercury species in these emissions have variable atmospheric residence times, which influence their atmospheric transport and deposition patterns. Forested regions with a prevalence of wetlands and of unproductive surface waters promote high concentrations of mercury in freshwater biota and thus are particularly sensitive to mercury deposition. Through fish consumption, humans and wildlife are exposed to methylmercury, which markedly bioaccumulates up the freshwater food chain. Average mercury concentrations in yellow perch fillets exceed the Environmental Protection Agency's human health criterion across the region, and mercury concentrations are high enough in piscivorous wildlife to cause adverse behavioral, physiological, and reproductive effects. Initiatives are under way to decrease mercury emissions from electric utilities in the United States by roughly 70%.
Mercury (Hg) is a potent neurotoxin of significant ecological and public health concern. Human and wildlife exposure to Hg occurs largely through the consumption of contaminated fish. It is estimated that over 410,000 children born each year in the United States are exposed in the womb to methylmercury (MeHg) levels that are associated with impaired neurological development (Mahaffey 2005). Eight percent of US women of childbearing age have blood Hg levels in excess of values deemed safe by the US Environmental Protection Agency (USEPA; Schober et al. 2003). Studies have also linked elevated Hg in the blood or tissue of fish, birds, and mammals with negative effects such as reduced reproductive success, hormonal changes, and motor skill impairment (Wiener and Spry 1996, Nocera and Taylor 1998, Evers et al. 2004).
To protect human health, the USEPA set a fish tissue criterion for MeHg at 0.3 μg per g under section 304(a) of the Clean Water Act (USEPA 2001). Similar criteria for wildlife are under development or promulgation in several states (e.g., Maine, New York). As of 2004, fish consumption advisories regarding Hg contamination have been issued for 44 states, including 21 statewide advisories for fresh waters and 12 for coastal waters. These advisories represent more than 53,000 km2 of lakes and 1,230,000 km of rivers. The extent of fish consumption advisories underscores the extensive human and ecological health risk posed by Hg pollution.
Important sources of Hg to the environment include electric utilities, incinerators, industrial manufacturing, wastewater treatment plants, and improper disposal of consumer products (e.g., batteries, fluorescent light bulbs, Hg switches). Considerable public policy attention is directed toward airborne Hg emissions, since they constitute the largest source of Hg in the United States and globally (UNEP 2002). Although estimates suggest that US emissions of Hg peaked in the 1970s and have since declined (Pirrone et al. 1998), atmospheric concentrations remain approximately three times higher than preanthropogenic levels (Mason et al. 1994).
Neither atmospheric Hg emissions nor ambient concentrations of Hg in water constitute a direct public health risk at the levels of exposure usually found in the United States. The risk to humans and wildlife occurs as Hg is transported to watersheds and accumulates in the aquatic food chain. Airborne Hg is transported over variable distances (i.e., local to global scales), depending on the speciation of Hg emissions and reaction pathways, and is deposited to the Earth's surface.
Following deposition, ionic Hg (i.e., oxidized mercuric species, including complexes and particulate forms) may be reduced and reemitted to the atmosphere or converted to a more bioavailable form, MeHg. Through a bioaccumulation factor of about 10 million, MeHg accumulates to toxic levels at the top of the aquatic food chain. This Hg linkage, from air to water to fish and other biota, challenges the state and federal regulators charged with controlling airborne emissions and with decreasing Hg deposition to levels that meet standards for concentrations in water and in fish tissue.
To improve understanding of the Hg air–water–biota connection, the Hubbard Brook Research Foundation convened a team of eight scientists to synthesize scientific information concerning (a) Hg sources and inputs; (b) Hg transport, transformations, exposure, and environmental effects; and (c) Hg policy impacts in the Northeast. This synthesis includes the analysis of a large Hg data set compiled for eastern North America as part of a NERC (Northeastern Ecosystem Research Cooperative) initiative (Evers and Clair 2005). The NERC Hg project published summaries for water, sediment, and major taxonomic groups. Here we distill these studies into a regional overview with policy applications.
Efforts have been under way at state, regional, national, and global scales to reduce Hg emissions. Notably, in May 2005 the USEPA adopted a rule pertaining to Hg emissions from coal-fired power plants (the Clean Air Mercury Rule, or CAMR). This rule calls for a two-phase reduction in emissions through a cap-and-trade approach that is predicted to produce by approximately 2025 a 70% decrease in total US emissions from electric utilities. Rather than imposing an emission rate limit or requiring the use of maximum achievable control technology, the cap-and-trade approach allows facilities to purchase Hg allowances in order to comply with the regulations.
Mercury emissions and deposition in the northeastern United States
The northeastern United States (i.e., New England and New York) is an important region in which to investigate Hg, because it receives elevated Hg deposition and contains ecosystems sensitive to Hg inputs. Mercury-sensitive areas are typically forested areas with shallow surficial materials, abundant wetlands, and low-productivity surface waters. In the Northeast, the fish in many lakes and streams and the associated wildlife have elevated Hg, which in some instances is high enough to constitute a “biological Hg hotspot,” which requires special attention from both a scientific and a policy perspective (Evers et al. 2007). A biological Hg hotspot is a location on the landscape that, compared with the surrounding landscape, is characterized by elevated concentrations of MeHg in biota (e.g., fish, birds, mammals) in excess of established human health or wildlife criteria as determined by a statistically adequate sample size.
Mercury emissions
Globally, approximately 6600 metric tons of Hg are emitted to the atmosphere annually, with 33% to 36% attributed to direct anthropogenic emissions. The remainder originates from natural sources or from past anthropogenic emissions that are rereleased (Mason and Sheu 2002). These values suggest that about two-thirds of atmospheric Hg emissions are derived from either direct or reemitted anthropogenic sources. Coal-fired power plants are the largest single category of Hg emissions, with 1450 metric tons per year, comprising about 50% of anthropogenic sources (Pacyna et al. 2003).
Total anthropogenic Hg emissions from all sources in the United States are calculated to be 103 metric tons per year, with the Northeast contributing about 4.7 metric tons per year (USEPA 1999). Mercury emissions in the United States have declined markedly over the past decade (table 1) as a result of federal regulations that mandated large reductions in Hg emissions in medical waste incinerators and in municipal incinerators (USEPA 2005). Unlike incinerator emissions, emissions from electric utilities have remained largely unchanged, and their relative contribution to total US emissions has increased from 25% to 40%. Municipal waste incinerators (23%) and electric utilities (16%) are the largest point-source categories in the Northeast.
Table 1.
Mercury (Hg) emissions (in metric tons per year), by source category, in the United States from 1990 through 2002 and in the Northeast region in 2002.

Mercury is emitted to the atmosphere from point sources in three forms: elemental Hg (Hg0), gaseous ionic Hg (reactive gaseous mercury, or RGM), and particulate Hg (PHg). This speciation exerts significant control over the fate of atmospheric Hg emissions and varies widely among sources (table 2). Therefore, Hg can be a local, regional, or global pollutant, depending on the speciation of the emissions and the associated residence times in the atmosphere (Dastoor and Larocque 2004).
Table 2.
Percentage of mercury species emitted, by source category.

In 1999, 57% of calculated point-source Hg emissions in the Northeast occurred as Hg0, 33% as RGM, and 10% as PHg (USEPA 1999). Studies indicate that emissions from coal combustion in the United States are roughly 50% Hg0, 40% RGM, and 10% PHg (Pacyna et al. 2003). However, emissions from coal combustion in the northeastern states have a higher percentage of RGM (68%) and a lower percentage of Hg0 (30%) and PHg (2%; NESCAUM 2005). The actual Hg emission speciation profile for a specific power plant depends on the type of coal used and the air pollution control technology employed (NESCAUM 2003).
Elemental Hg, which is relatively unreactive and generally slowly oxidized, constitutes by far the largest pool of Hg in the atmosphere because of its relatively long residence time (0.5 to 2 years) and long-range transport potential (tens of thousands of kilometers). However, under some conditions Hg0 can be rapidly oxidized and deposited locally or regionally, as observations have shown in the Arctic and Antarctic (Lindberg et al. 2002), at the marine and continental boundary layer, and in areas downwind of urban areas (Weiss-Penzias et al. 2003). Elemental Hg can also be directly deposited to forested ecosystems through stomatal gas exchange (Grigal 2002). As a result, the atmospheric lifetime of Hg0 is probably closer to 0.5 year than to 2 years.
Reactive gaseous Hg consists predominantly of gaseous chloride and oxide forms of ionic Hg. This species is highly soluble in water and readily deposits to surfaces within tens to a few hundreds of kilometers from emission sources. Because of RGM's short atmospheric residence time (0.5 to 2 days), elevated Hg deposition can occur near RGM emission sources.
The atmospheric residence time of PHg is also relatively short (0.5 to 3 days). Although the fraction of PHg in ambient air in remote areas is generally less than 5% of total atmospheric Hg (Horvat 1996), concentrations may be higher near Hg emission sources and under certain atmospheric conditions (Lu et al. 2001).
Atmospheric deposition
Atmospheric deposition of Hg occurs in two forms: wet deposition (the deposition of Hg associated with rain and snow) and dry deposition (the deposition of PHg and RGM, cloud and fog deposition, and stomatal uptake of Hg0). Although some areas have been contaminated by land disposal of Hg or discharge of Hg in wastewater effluent, the predominant input of Hg to most watersheds is atmospheric deposition. Fitzgerald and colleagues (1998) systematically rule out alternate hypotheses, such as natural weathering, as a significant cause of the observed widespread Hg contamination.
Judging from global models (Hudson et al. 1995), reconstructions of mass balances (Mason et al. 1994), and paleo-limnological techniques (Engstrom and Swain 1997), it appears that deposition of Hg has increased two- to threefold over the past two centuries, following increases in Hg emissions associated with industrialization and Hg use. Paleo-limnological studies in the Northeast typically show Hg deposition starting to increase in the late 1800s or early 1900s and increasing 2.5- to 15-fold by the late 20th century (1970s to 1990s) (figure 1; Kamman and Engstrom 2002). Decreases in sediment Hg deposition in the Northeast (approximately 25%) have been evident in recent years, coincident with reductions in US emissions and with static global emissions. Because inventories of Hg emissions have been limited, it is not clear what is responsible for the declines in Hg deposition over the past few decades. However, it seems likely that controls on particulate matter and sulfur dioxide from electric utilities, and reductions in consumer and industrial Hg use, are important factors (Engstrom and Swain 1997).
Figure 1.
Changes in historical deposition of mercury (Hg) to sediments in (a) Spring Lake and (b) Wallingford Pond, Vermont, from 1820 to the present (after Kamman and Engstrom 2002). The sediment patterns reflect changes in Hg emissions and deposition over time.

In the eastern United States, Hg deposition is high (USEPA 1997), but it is difficult to identify its specific sources. Of the estimated 52 metric tons of Hg deposited per year in the United States from US sources, 24 metric tons (46%) are likely to originate from domestic utility coal boilers (half of the 48 metric tons of Hg that the coal-fired utilities emit each year is likely to be deposited within the United States; USEPA 1997). Likewise, for regions of New York it is estimated that 11% to 21% of the Hg deposited is derived from emissions within New York, 25% to 49% originates from other US sources, and 13% to 19% originates from Asia (Seigneur et al. 2003). Given that most coal-fired utilities emit 50% to 70% of Hg as RGM and PHg (table 2), local sources are most likely an important component of the deposition in areas within 50 km of these sources. An analysis of emissions and deposition in southern New Hampshire shows a local region of high deposition associated with local electric utility emissions (Evers et al. 2007).
In the United States and Canada, measurements of wet Hg deposition, which are largely made through the Mercury Deposition Network (MDN), show that wet Hg deposition is highest in the Southeast (e.g., Florida, Mississippi) and lowest in the West. There are currently seven MDN sites in the Northeast, with average annual wet deposition ranging from 3.8 to 12.6 μg per m2 per year ( http://nadp.sws.uiuc.edu/mdn/). There do not appear to be broad spatial patterns in wet Hg deposition across the region, but the network is sparse. Because of the placement of collectors in rural areas, the deposition values for the region do not include elevated deposition that would be expected near Hg sources and in urban areas.
Estimates of dry Hg deposition are highly uncertain because of the complex interrelationships of atmospheric conditions, collection surface characteristics and terrain, and chemical properties of the contaminants. Several modeling efforts have been used to estimate dry deposition of Hg, however. In regions of New York, estimated dry Hg deposition was 4 to 10 μg per m2 per year (Seigneur et al. 2003). Another model estimate specifically for the Northeast suggests that dry deposition of RGM plus Hg0 was 37 μg per m2 per year (Xu et al. 2000). Both studies indicate that dry deposition provides a significant pathway of Hg inputs (50% to 75% of total deposition) and agree with USEPA predictions that Hg dry deposition in the Northeast is the highest in the country, in part as a result of the abundant forests whose canopies effectively collect Hg from the atmosphere.
Because of the large surface area associated with canopy foliage, atmospheric deposition of contaminants is elevated in forests compared with other types of ecosystems. Forest studies have indicated that total atmospheric Hg deposition may be estimated using fluxes of throughfall (precipitation that passes through the canopy) plus litterfall (plant material that falls to the forest floor; Rea et al. 2001). Grigal (2002) suggests that the ratio of Hg fluxes resulting from wet deposition, throughfall, and litterfall, respectively, is 1.0 to 1.8 to 2.2. So for the 5 μg per m2 per year of wet deposition that might be typical of the Northeast, anticipated through-fall would be 9 μg per m2 per year, and litterfall would be 11 μg per m2 per year, resulting in total Hg deposition of 20 μg per m2 per year and dry deposition of 15 μg per m2 per year (75% of total).
Some portion of the Hg deposited to Earth's surface is reemitted to the atmosphere. However, rates of volatilization vary widely in association with differences in vegetation, soil moisture, temperature, solar radiation, and landscape characteristics. In general, volatilization rates from soil are high immediately after inputs of ionic Hg to the soil (Schluter et al. 1995). On the basis of a review of the literature, Grigal (2002) estimated a mean rate of Hg0 volatilization from soil of approximately 11 μg per m2 per hour. This rate is more than adequate to reemit most of the atmospheric Hg deposition. The magnitude and uncertainty of this process demonstrate the acute need for additional research on Hg reemissions.
Transport and transformation of mercury in forest–wetland–lake ecosystems
Following deposition to the landscape, Hg may be sequestered in soil, reemitted to the atmosphere, or transported through the watershed, with a fraction of these inputs ultimately supplied to surface waters. Watershed and water chemistry characteristics influence the transport of Hg to surface waters. Anoxic zones in wetlands and lakes provide suitable conditions for the methylation of ionic Hg to MeHg. The extent to which MeHg is biomagnified in the freshwater food chain depends on the nature and length of the food chain and on water chemistry characteristics.
Mercury transport and fate in upland forest ecosystems
Although there have been few direct studies of soil sequestration of Hg, immobilization of Hg in forest soil is known to correspond with the retention of organic carbon (Schwesig et al. 1999). Pools of Hg in upland soil in northern temperate regions are about 7 mg per m2, although higher levels have been reported in central Europe (Grigal 2003).
The export of Hg by waters draining upland soils to surface waters is generally low. Concentrations and fluxes of Hg in soil waters, as in soil, are closely related to dissolved organic carbon (DOC; Schwesig et al. 1999). In northern forests, concentrations of total Hg are highest in waters draining the upper soil, coinciding with high concentrations of DOC. Concentrations and fluxes of total Hg decrease as DOC is immobilized with depth in mineral soil (Grigal 2002).
Limited studies suggest that MeHg concentrations in upland soils and groundwaters are generally low, although higher concentrations occur in upper soil waters and decrease with soil depth (Grigal 2002). Low concentrations and fluxes of MeHg in drainage waters suggest that rates of methylation are low, and freely draining upland soils are generally not important in the supply of MeHg to downstream surface waters, with the possible exception of recently harvested forests (Porvari et al. 2003).
Transport and transformation of mercury in wetlands
Wetlands are important features of the landscape that influence the supply of different Hg species to adjacent surface waters. Wetlands are typically net sinks of total Hg and sources of MeHg (Grigal 2002, 2003). Rates of total Hg accumulation are greater in wetlands than in upland soils because of the strong association of Hg with organic matter (Grigal 2003). Annual rates of MeHg production in wetlands are approximately 0.1 to 1 μg per m2 per year (Galloway and Branfireun 2004). The factors controlling methylation of Hg in wetlands are not completely understood, but they most likely involve the amounts and types of organic matter, hydrologic flow paths, and rates of microbial activity (Galloway and Branfireun 2004). Wetlands are also a major source of DOC. Organic matter produced in wetlands forms complexes with both ionic Hg and MeHg, enhancing the transport of these Hg species to surface waters but decreasing their bioavailability (Hudson et al. 1994). An elevated supply of DOC to downstream surface water could also stimulate methylation and limit photodegradation of MeHg and photoreduction of ionic Hg. Furthermore, wetlands support sulfate-reducing bacteria, which appear to be largely responsible for Hg methylation (Benoit et al. 2003). Concentrations of MeHg in wetland porewaters (waters filling the spaces between solid material in sedimentary deposits) and surface waters vary seasonally, with the highest concentrations evident during the late summer, presumably as a result of warmer temperatures, higher rates of microbial activity, and longer hydraulic residence times (Galloway and Branfireun 2004).
Mercury concentrations and transformations in surface waters
Freshwater ecosystems are among the most sensitive to Hg pollution. Total Hg concentrations in surface waters in the Northeast vary by more than an order of magnitude, from less than 0.5 to 12.7 nanograms per liter (5th to 95th percentile; figure 2; Dennis et al. 2005). Most of the Hg in surface water occurs as ionic Hg, with MeHg ranging from 1% to 35% of total Hg (figure 3). Under conditions of high total Hg loading, MeHg production can vary widely, depending on the methylation efficiency of a particular ecosystem (Krabbenhoft et al. 1999).
Figure 2.
Average water mercury (Hg) concentrations within 18-minute grid cells for lakes and streams across northeastern North America. Inset shows the distribution of Hg concentrations comprising the mean for each quintile.

Figure 3.
Box and whisker plots of mercury (Hg) concentrations in water and aquatic biota in eastern North America. Also shown are the ranges for the percentage of total Hg occurring as methylmercury (MeHg). All values were obtained from NERC (Northeastern Ecosystem Research Cooperative) data and represent wet weight, except those for phytoplankton, which were obtained from Watras and colleagues (1998).

Mercury enters remote surface waters through direct atmospheric deposition and through soil water, wetland, or groundwater drainage. Streams and rivers can exhibit marked temporal variation in Hg concentrations, which is associated with variations in concentrations of DOC or suspended matter. Large increases in Hg concentrations can occur during high flow events (Shanley et al. 2005).
Some inputs of Hg to lakes are removed from the water column by the volatilization of Hg0 and by sediment deposition. In freshwater lakes, photochemical processes are largely responsible for the reduction of ionic Hg to Hg0 (Amyot et al. 1997). Microbial reduction has been observed in laboratory studies, but only at higher than ambient concentrations of Hg (Morel et al. 1998). Biogeochemical processes in lakes also result in net production of MeHg due to methylation in anoxic sediments and in the water column.
The geographic distribution of average surface water Hg concentrations in the Northeast (figure 2) shows landscape-level heterogeneity in lake and river Hg concentrations, and areas where concentrations are elevated across several contiguous 18-minute grid cells. Areas of elevated Hg concentrations in surface waters can be explained by high concentrations of DOC, as in the Adirondacks; by high inputs of suspended solids, from rivers along Lake Champlain, related to high flow events; and by elevated atmospheric Hg deposition, as in lakes in southeastern New Hampshire and eastern Massachusetts. A large portion of the variation in total Hg and MeHg across the region can be explained by variation in DOC (Dennis et al. 2005). Areas with the highest mean surface water Hg concentrations also have the greatest range in Hg concentrations (figure 2). This variation may be attributed to heterogeneity in watershed characteristics or to high flow events (Shanley et al. 2005).
Other factors controlling mercury dynamics in surface waters
Other factors, such as water chemistry, land cover and land use, and watershed disturbances, alter the transport, transformation, and bioavailability of Hg in surface waters.
The Northeast receives elevated loading of acidic deposition as well as Hg deposition, and contains a relatively large number of acidified surface waters. Acidic deposition and the associated sulfur alter the acid–base status of surface waters, thereby influencing Hg transformation and accumulation in fish. Sulfur transformations are closely coupled with Hg dynamics. The solubility of Hg increases with increasing sulfide concentrations in anoxic waters through complexation reactions, potentially increasing the pool of Hg available for methylation (Benoit et al. 2003). Experimental observations show that when sulfate is added to wetlands or lakes, sulfate reduction is enhanced, leading to increased methylation and MeHg export (Branfireun et al. 1999, Watras et al. 2006).
Widespread observations show an inverse relationship between fish Hg concentrations and surface water pH (e.g., Kamman et al. 2004). Hrabik and Watras (2002) used reference data and observations from a lake experimentally acidified with sulfuric acid to examine the relative contribution of atmospheric Hg deposition and acidic deposition to Hg concentrations in fish. They found that half of the decrease in fish Hg over a six-year period during which the lake was recovering from acidification could be attributed to decreases in sulfuric acid loading.
In a study of 21 river basins nationwide, watersheds with mixed agriculture and forest land cover had the highest methylation efficiency, even where these watersheds had low total Hg in sediments (Krabbenhoft et al. 1999). Some waters draining largely agricultural lands have relatively high concentrations of total Hg and MeHg, but lower concentrations in fish, presumably due to algal “bloom dilution” associated with high phosphorus loading (Kamman et al. 2004; see below) or elevated DOC concentrations (which could stimulate methylation but limit bioaccumulation), or both.
Land disturbance influences Hg export and availability for methylation. Forest harvesting has been shown to increase export of total Hg and MeHg (Porvari et al. 2003). Fire results in a complex pattern of Hg loss from watersheds. During and shortly after fire, elevated Hg losses are associated with volatilization and drainage losses (Grigal 2002). Over the longer term, Hg transport to surface waters is reduced in burned areas as a result of decreases in soil carbon and DOC concentrations.
In reservoirs, rates of Hg methylation can be altered by water level fluctuation associated with hydropower production or flood control. Many large bodies of water in the Northeast are impounded to increase their storage or daily peaking capacity, and these water bodies may fluctuate tens of centimeters on a daily basis or several meters over the course of a summer. As the littoral zone experiences periodic wetting and drying, varying cycles of reduction and oxidation may enhance the production of MeHg, depending on a variety of factors (Sorensen et al. 2005, Evers et al. 2007).
Trophic transfer of mercury in surface waters of the Northeast
Concentrations of total Hg or MeHg in surface waters often do not correlate well with the Hg content of freshwater biota, such as fish. There are many physical, chemical, ecological, and land-use factors controlling the trophic transfer of MeHg, which are key to predicting MeHg concentrations in fish and other freshwater organisms.
Trophic transfer of Hg in freshwater food webs begins with the bioaccumulation of ionic Hg and MeHg by primary producers. Bioaccumulation factors in the transfer of Hg from water to algae are by far higher (approximately 105 to 106) than at subsequent trophic levels (figure 3). Although both ionic Hg and MeHg are taken up by aquatic organisms, MeHg is assimilated four times more efficiently than ionic Hg (Mason et al. 1994). However, the absolute and relative assimilation efficiencies of ionic Hg and MeHg vary with trophic level, uptake pathway, and water chemistry conditions. Freshwater grazers and predators acquire MeHg mainly from their food rather than from water (Harris and Bodaly 1998). Methylmercury is efficiently transferred to the higher levels of the food web and largely incorporated within proteins, as in muscle tissue.
The NERC data show that MeHg increases in concentration and comprises a greater percentage of the total Hg in freshwater consumers and predators as it progresses up the food chain (figure 3). Thus organisms consuming prey at higher trophic levels are exposed to higher concentrations of total Hg and MeHg (Vander Zanden and Rasmussen 1996). Fish Hg occurs almost entirely as MeHg.
A variety of physical, chemical, and biological factors influence the biomagnification of MeHg. Fish Hg concentrations tend to vary positively with lake or watershed area and negatively with pH, acid neutralizing capacity (ANC), nutrient concentrations, zooplankton density, and human land use (Chen et al. 2005). Furthermore, the Hg added to the lake surface each year appears to be more available for conversion to MeHg than Hg that has been in the ecosystem for longer periods (Gilmour et al. 2003).
Both experimental and field studies show that nutrient enrichment diminishes Hg bioaccumulation in phytoplankton through the biodilution of Hg under algal bloom conditions (Pickhardt et al. 2002). Mercury concentrations in zooplankton also decrease with increasing zooplankton densities that in turn are correlated with lower Hg concentrations in fish (Chen and Folt 2005). Growth dilution in fish, also under conditions of high productivity and food availability, may be related to lower Hg concentrations in fish (Essington and Houser 2003).
Within given fish populations, Hg burdens increase with the age and size of individuals in part because of the slower rates of elimination and longer exposure in larger individuals, and in part because of the consumption of higher-trophic-level foods by older and larger individuals (Wiener and Spry 1996). Mercury concentrations in top predator fish are higher in food webs with longer chain lengths and less omnivory (Stemberger and Chen 1998).
Indicators of mercury sensitivity
Four simple and common measures of water quality—DOC, ANC, pH, and total phosphorus—have been shown by Chen and colleagues (2005) and many others to be related to fish Hg concentrations. To develop indicators of Hg sensitivity, we combined data from two stratified, random-probability surveys of northeastern lakes (USEPA EMAP [Environmental Monitoring and Assessment Program], Northeast Lakes Program, 1991–1994, and Vermont–New Hampshire REMAP [Regional EMAP], 1998–2000) with the survey data sets of Chen and colleagues (2005) to examine these four water-chemistry characteristics in lakes with standard-age yellow perch (Perca flavescens) whose tissue contained mean concentrations of Hg above and below the USEPA criterion (0.3 μg per g; figure 4). The standard age for yellow perch examined in this analysis was 4.6 years (Kamman et al. 2004). This analysis showed that lakes with Hg levels above 0.3 μg per g in yellow perch had significantly higher DOC (t = −3.099, p = 0.003) and lower pH (t = −6.282, p < 0.001), ANC ( t = 2.835, p = 0.007), and total phosphorus (t = 3.840, p < 0.001) than lakes with fish Hg concentrations below 0.3 μg per g. As yellow perch have low to moderate Hg concentrations, these thresholds are conservative and help identify the most sensitive lakes.
Figure 4.
Relationship between methylmercury (MeHg) concentrations in standard-length yellow perch and total phosphorus concentration in lakes (a), and box and whisker plots of concentrations of dissolved organic carbon (b), pH (c), and acid neutralizing capacity (d) for lakes in the northeastern United States containing average concentrations of standard-age yellow perch with MeHg concentrations less than and greater than 0.3 μg per g.

Twenty percent of lakes in the region had total phosphorus concentrations above 30 μg per L. In those lakes, Hg concentrations in yellow perch were below 0.3 μg per g. In the remaining 80%, we found that most lakes (75%) had yellow perch Hg concentrations exceeding 0.3 μg per g when surface waters had a DOC level of more than 4.0 mg carbon per L, a pH of less than 6.0, or an ANC of less than 100 micro-equivalents (μeq) per L. These commonly monitored indicators provide natural resource managers with a useful tool for evaluating the likelihood of high fish Hg concentrations in individual lakes.
Taxonomic patterns of mercury exposure
Biota are exposed to MeHg primarily through fish and insect consumption. The NERC data establish robust Hg exposure profiles for fish, birds, and mammals (table 3; Evers and Clair 2005), and highlight the importance of habitat type, foraging guild, trophic structure, and demographics on MeHg exposure (Evers et al. 2005).
Table 3.
Mercury exposure for selected biota in representative habitats in the Northeast.

In general, Hg concentrations vary by taxonomic group, with a higher proportion of MeHg at higher trophic levels. Mercury in benthic invertebrates and larval insects has been extensively studied in northeastern lakes and reservoirs, and is found to increase with trophic level (odonates > hemipterans and coleopterans > trichopterans > dipterans and ephemeropterans; Tremblay et al. 1996). The NERC data on Hg in over 15,000 fish show that the mean fillet Hg levels in 10 of the 13 species are above 0.3 μg per g, with the highest levels in large predatory fish such as walleye (Sander vitreus) and lake trout (Salvelinus namaycush; figure 5; Kamman et al. 2005).
Figure 5.
Mean and standard deviation of mercury (Hg) concentrations of 13 species of fish in eastern North America (Kamman et al. 2005). The downward-pointing arrow indicates the US Environmental Protection Agency's criterion for fish Hg concentrations.

Habitat type also has an important influence on MeHg concentrations. Data for two-lined salamanders (Eurycea bislineata) suggest that amphibians found in headwater streams have significantly higher MeHg concentrations than those in lakes (Bank et al. 2005). Larval insects in reservoirs have total Hg concentrations that are 3 to 10 times higher than those in natural lakes (Tremblay et al. 1996). Northern crayfish (Orconectes virilis) in headwater streams have Hg concentrations up to five times greater than those in lakes (Pennuto et al. 2005).
Comprehensive bird studies illustrate differences in MeHg exposure in foraging guilds. Piscivorous species with particularly high MeHg levels include the common loon (Gavia immer; Evers et al. 2005), wading birds (Frederick et al. 1999), and the bald eagle (Haliaeetus leucocephalus; Bowerman et al. 2002). Exposure studies in common loons have shown hormonal changes, reduced reproductive success, and motor skill impairment, resulting in the establishment of a wildlife criterion for blood Hg of 3.0 μg per g (Evers et al. 2004). Exposure to MeHg is not limited to piscivorous birds. Data for insectivorous songbirds, such as the northern waterthrush (Seiurus noveboracensis) and red-winged blackbird (Agelaius phoeniceus), show blood Hg levels that can exceed levels in piscivorous birds (Evers et al. 2005). Moreover, elevated MeHg has been measured in several breeding populations of saltmarsh sharp-tailed sparrows (Ammodramus caudacutus) in some New England estuaries (Lane and Evers 2005), and in terrestrial species such as Bicknell's thrush (Catharus bicknelli) and other montane songbirds (Rimmer et al. 2005).
Terrestrial mammals, particularly mink (Mustela vision) and river otter (Lontra canadensis; table 3), also experience elevated MeHg in the Northeast. Yates and colleagues (2005) found that Hg levels tend to be higher in mink than in otter, in interior than in coastal populations, and in females than in males. Recent evidence for MeHg exposure in insectivores has led to ongoing investigations in bats and other nonpiscivorous mammal species.
Comprehensive data on fish and wildlife exposure are being used to identify species, habitats, and regions that are likely to be at the highest risk for MeHg contamination, and will be useful for measuring progress resulting from future management actions.
Evaluating reductions in mercury emissions
At present, most state and national policy attention is focused on Hg emissions from electric utilities (i.e., coal-fired power plants). Although controlling other sources (e.g., emissions from incinerators, discharges from wastewater treatment plants) and implementing other management options (e.g., biomanipulation, land-use management) may also hold promise for reducing and mitigating Hg bioaccumulation, we focus on the potential effect of reducing Hg emissions from electric utilities, because they are the largest single source of airborne emissions in the United States and the second largest source in the Northeast, and because their emissions have remained unchanged both regionally and nationally over the past decade (NESCAUM 2005). Although municipal waste combustors are the largest Hg emission source in the Northeast, effective strategies for reducing their emissions are under way, as evidenced by the decline of approximately 80% in emissions from this source between 1998 and 2003 (NESCAUM 2005).
Many proposals have been introduced at both the federal and the state level to control Hg emissions from electric utilities. The main differences among them include (a) the level and timing of the cuts, (b) the existence of an emissions cap or emissions rate limit, and (c) whether or not trading is allowed. In general, the level and timing of Hg emission reductions are likely to control the extent and rate of recovery in the region, and the use of trading has prompted questions regarding the persistence or expansion of biological Hg hotspots (Evers et al. 2007).
Here we estimate the changes in emissions and deposition that are associated with the CAMR and discuss the potential effect of these changes on freshwater ecosystems using field data. The USEPA estimates that the CAMR will result in a 70% decrease in Hg emissions from electric utilities by 2025. We estimate that the CAMR, when fully implemented, would result in a decrease of approximately 18% to 30% in deposition in the northeastern United States. This estimate is based on an analysis of US emissions and deposition that assumes (a) that current and reemitted anthropogenic emissions each constitute one-third of the emissions in the United States, and (b) that electric utilities account for 50% of each of these two emission categories. It follows that if electric utilities reduce their emissions by 70%, current and reemitted anthropogenic emissions would each decrease by 35%.
We further assume that US emissions are responsible for 40% to 65% of Hg deposition in the Northeast (Seigneur et al. 2003) and that reemitted US emissions contribute another 10% to 20%. If deposition attributed to these emission categories were reduced by 35% as a result of the CAMR, then total deposition would decline by approximately 18% to 30%. These predictions are consistent with the decrease of approximately 25% in sediment Hg deposition that occurred coincident with decreases in Hg emissions in the United States between 1970 and 1999.
An 18% to 30% decrease in Hg deposition is likely to provide significant ecological benefits in the region. Detailed biological data from a group of nine lakes in New Hampshire show that the Hg concentrations in the blood and eggs of the common loon declined 50% between 1999 and 2002 as emissions in the vicinity were cut 45% between 1997 and 2002, suggesting that some ecosystems in close proximity to large emissions sources may experience rapid improvement (Evers et al. 2007). Hrabik and Watras (2002) found that Hg fish concentrations declined 30% between 1994 and 2000 as a result of decreased atmospheric Hg loading to a lake in northern Wisconsin; they concluded that modest changes in Hg or acidic deposition can significantly affect Hg bioaccumulation over short timescales. The range and rate of ecosystem response are most likely related to the variation in the physical, chemical, and biological characteristics of lakes and watersheds.
We expect that the CAMR will produce important results, but these changes may not be sufficient to protect human and environmental health. Given that average fish Hg concentrations sampled across the region currently exceed the USEPA human health criterion by 10% to 88%, depending on the species, significant additional reductions in Hg emissions from other US and global sources will probably be necessary to bring about widespread recovery to Hg levels that are below this criterion in most fish species in the northeastern United States.
Conclusions
A large Hg database produced by the NERC Hg working group was used to document and examine the widespread Hg contamination across eastern North America. From this synthesis, it is evident that the Northeast receives elevated Hg deposition derived mostly from direct emissions and re-emissions of anthropogenic sources. Paleolimnological studies suggest that Hg deposition is substantially influenced by US emissions and responds to reductions in these sources.
Direct anthropogenic emissions of Hg originate largely from electric utilities, incinerators, and industrial processes. Current understanding of speciation and deposition processes suggests that, while speciation exerts important influence over patterns of atmospheric transport and deposition, all forms of Hg have the potential to deposit locally or regionally.
Forest regions are particularly sensitive to Hg inputs as a result of numerous factors: the filtering effects of the canopy and the associated elevated deposition; the prevalence of wetlands, which are critical in the transport of Hg and the production of MeHg; and low-productivity lakes, which promote high concentrations of Hg in fish. Although Hg is highly variable in surface waters across the region, we have identified several chemical thresholds to predict high fish Hg: total phosphorus concentrations of less than 30 μg per L; pH of less than 6.0; ANC of less than 100 μeq per L; and DOC of more than 4 mg carbon per L. Freshwater food chains are characterized by marked bioaccumulation of MeHg (106 to 107), with the largest increase occurring from water to plankton (105). Many freshwater and terrestrial animals in the Northeast exhibit high concentrations of Hg. For the common loon, existing Hg concentrations can cause adverse individual (behavioral and reproductive) and population-level effects.
Our analysis suggests that (a) cuts in Hg emissions from electric utilities in the United States will decrease Hg deposition in the region; (b) decreased Hg deposition will result in lower Hg levels in biota, although significant time lags may exist in many ecosystems; and (c) widespread recovery to Hg levels that no longer pose a human health risk or population risk to the common loon will be a long-term process that is likely to require additional reductions in Hg emissions.
Acknowledgments
This work was convened through the Science Links program of the Hubbard Brook Research Foundation with support from the Henry Luce Foundation, the John Merck Fund, the Merck Family Fund, the Harold Whitworth Pierce Charitable Trust, the New York Energy Research and Development Authority, and the Syracuse Center for Environmental and Energy Systems. This project was also supported through grants from the US Environmental Protection Agency (USEPA) and the National Science Foundation to C. T. D. and the National Institute of Environmental Health Sciences (NIH grant no. P42 ESO7373) to C. Y. C. The Hubbard Brook Experimental Forest is operated and maintained by the Northeastern Research Station, US Department of Agriculture, Newton Square, Pennsylvania. We would particularly like to thank Edward Swain (Minnesota Pollution Control Agency) for serving as an advisor to this project. We also thank several anonymous reviewers. The findings published here are independent and do not necessarily reflect the views of the advisors.