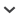The superfamily Certhioidea is distributed on four continents and while comprising relatively few species, includes forms as diverse as creepers, nuthatches, gnatcatchers, and wrens. Previous attempts to infer the phylogeny of this lineage have focused on its higher-level relationships, consequently undersampling the New World wrens. This study reports the first nearly genus-level sampling of certhioids, based on concatenated and species tree analyses of 8520 bases of DNA sequence data from six gene regions. These analyses, while failing to completely resolve basal certhioid relationships, corroborate the monophyly of a diverse New World clade of gnatcatchers, gnatwrens, and wrens, and significantly improve our understanding of wren relationships. The inferred relationships among certhioids and wrens support an Old World origin for these lineages, with dispersal of the New World clade in the mid-Miocene, suggesting expansion and early diversification of the lineage through North America. This scenario suggests a minimum of six independent dispersal events into South America in this lineage, at least some likely to have been made prior to the Pliocene.
INTRODUCTION
Previous Hypotheses of Relationship within Certhioidea
The superfamily Certhioidea is a small clade of passerine birds comprising the families Sittidae (nuthatches and the wallcreeper), Certhiidae (creepers), Polioptilidae (gnatcatchers and gnatwrens), and Troglodytidae (wrens). The monophyly of this group of approximately 141 species (Gill and Donsker, 2017) was first indicated by analyses of DNA-DNA hybridization data (Sibley and Ahlquist, 1990; fig. 1). These analyses also suggested the group was most closely related to Old World warblers, bulbuls, titmice, and relatives, leading to its placement within the oscine passerine superfamily Sylvioidea. Subsequent analyses of family-level passerine nuclear sequence data questioned placement in Sylvioidea, instead suggesting recognition at the superfamily level (Barker et al., 2002; Barker et al., 2004; Cracraft et al., 2004). More recent molecular work has consistently supported monophyly of members of this clade (Alstrom et al., 2006; Johansson et al., 2008; Fregin et al., 2012; Moyle et al., 2016; Zhao et al., 2016), although to date few studies have included representatives of all its major lineages.
As for most passerine families, this superfamily has no known morphological synapomorphies. That the close relationship of species in the group was not previously recognized is likely due to the fact that it comprises at least four major ecotypes with differing locomotory and feeding specializations that do not appear entirely concordant with phylogeny. In the hind limb, the group is split between strong graspers that can cling to branches, vertical trunks, or even sheer stone surfaces (nuthatches, the wallcreeper, and some wrens) and relatively weak graspers that cling to trunks with tail support (creepers) or are limited to branches or horizontal surfaces (gnatcatchers, gnatwrens, and some wrens). In the rostrum, the group is split among species with long narrow mandibles specialized for reaching prey in crevices (creepers, the wallcreeper, gnatwrens, and most wrens), those with short broad bills that are used to excavate prey and even nest cavities (nuthatches), and those with delicate surface gleaning bills (gnatcatchers and some wrens). This morphological and ecological diversity led early taxonomists to associate these groups with many convergently similar lineages, including the Australasian sittellas (Daphoenositta) and treecreepers (Climacteris), the Philippine creeper (Rhabdornis), the Malagasy coral-billed nuthatch (Hypositta), and the Old World warblers (Sylviidae sensu lato, now Sylviidae, Acrocephalidae, Phylloscopidae, etc.; Alstrom et al., 2006; Fregin et al., 2012).
By contrast, relatively little consensus has been reached regarding relationships within the group. Perhaps the strongest single result has been monophyly of the New World lineage of Certhioidea, comprising the Polioptilidae and Troglodytidae. Every study including both lineages (e.g., Sibley and Ahlquist 1990; Barker et al., 2004; Barker 2004; Alstrom et al., 2006; Johansson et al., 2008; Zhao et al., 2016) has found strong support for their sister-group relationship, although Sibley and Ahlquist (1990) found the New World verdin (Auriparus) sister to the Polioptilidae, apparently due to a lab error or sample misidentification (subsequent studies have found Auriparus related to the penduline tits; e.g., Sheldon and Gill, 1996; Johansson et al., 2008). The most consistently sampled lineages other than the wrens and gnatcatcher clade (the New World Certhioidea, NWC) have been creepers (Certhia) and nuthatches (Sitta). All three possible relationships among these two genera and the New World lineage have been recovered in various studies: NWC+Certhia (Barker et al., 2002; Moyle et al., 2016; Zhao et al., 2016), NWC+Sitta (Alstrom et al., 2006; Johansson et al., 2008), and Sitta+Certhia (Barker et al., 2004). In terms of number of data sets (the three noted above) and data set size (4155 loci in Moyle et al., 2016), the evidence appears to be in favor of a NWC+Certhia relationship. Placement of the enigmatic genera Salpornis (the spotted creeper) and Tichodroma (the wallcreeper) relative to the NWC and Certhia remains somewhat in question: only three studies to date have included the former and only two the latter. The three studies including Salpornis have placed it either as sister to Sitta (Johansson et al., 2008) with weak support (although with strong support separating Salpornis from Certhia), or as sister to Certhia (Sibley and Ahlquist, 1990; Zhao et al., 2016) with strong support (or unevaluable support, in the case of DNA-DNA hybridization tree). The two studies to date that have placed Tichodroma phylogenetically (Sibley and Ahlquist, 1990; Zhao et al., 2016) have supported its relationship with Sitta (with strong support in the latter study), as expected by previous morphological and behavioral evaluations (reviewed in Vaurie, 1957; Sibley and Ahlquist, 1990). Excepting the placement of Auriparus, the current consensus on certhioid relationships looks essentially the same as that of Sibley and Ahlquist in 1990 (fig. 1).
Hypotheses of Relationship within Troglodytidae
The wrens are the most diverse lineage of certhioids, comprising at least 84 species (60% of the superfamily) in 19 genera (Gill and Donsker, 2017). Despite this diversity, to date only a few studies have addressed higher-level relationships within the group in any detail. Sibley and Ahlquist sampled only eight species in as many genera (fig. 1), and found relatively little structure among them, with Microcerculus falling out as most divergent, and a close relationship between the Carolina (Thryothorus ludovicianus) and Bewick's (Thryomanes bewickii) wrens. Overall divergence within wrens had a maximum of  T50H = 6.0, roughly suggesting a clade age of ~14 Ma (assuming 2.35 Ma/T50H, half the value for nonpasserines; Sibley and Ahlquist, 1990). More recent work on wren relationships based on DNA sequence data has improved our understanding of wren relationships (summarized in fig. 2). Barker (2004) showed: (1) the root of the wren tree most likely lay among a grade of highly terrestrial wrens including Salpinctes, Catherpes, Hylorchilus, and Microcerculus; (2) a close relationship between the genera Cistothorus and Troglodytes; (3) a close relationship between Campylorhynchus and Thryomanes/Thryothorus ludovicianus; (4) paraphyly of Thryothorus as recognized at the time; and (5) a well-supported relationship of “Thryothorus” except the type (T. ludovicianus) with the wren genera Cyphorhinus, Henicorhina, Uropsila, and Cinnycerthia. Subsequent work by Mann et al. (2006) extending sampling of “Thryothorus” to nearly all species of the group corroborated previous results and showed that all members of the genus except the type fell into three major clades more closely related to other genera than to the type. Two of those clades had available generic names that were resurrected, and Mann et al. erected a new genus for the third. Additional work on the genus Troglodytes and allies (Rice et al., 1999; Gómez et al., 2005) has shown that: (1) the Timberline Wren (Thryorchilus browni) is a close relative of the genus, (2) the Winter Wren may best be recognized in its own genus, Nannus (although evidence for its exclusion from Troglodytes is not overwhelming); and (3) the Socorro Wren, traditionally placed in Thryomanes, is actually a member of Troglodytes.
T50H = 6.0, roughly suggesting a clade age of ~14 Ma (assuming 2.35 Ma/T50H, half the value for nonpasserines; Sibley and Ahlquist, 1990). More recent work on wren relationships based on DNA sequence data has improved our understanding of wren relationships (summarized in fig. 2). Barker (2004) showed: (1) the root of the wren tree most likely lay among a grade of highly terrestrial wrens including Salpinctes, Catherpes, Hylorchilus, and Microcerculus; (2) a close relationship between the genera Cistothorus and Troglodytes; (3) a close relationship between Campylorhynchus and Thryomanes/Thryothorus ludovicianus; (4) paraphyly of Thryothorus as recognized at the time; and (5) a well-supported relationship of “Thryothorus” except the type (T. ludovicianus) with the wren genera Cyphorhinus, Henicorhina, Uropsila, and Cinnycerthia. Subsequent work by Mann et al. (2006) extending sampling of “Thryothorus” to nearly all species of the group corroborated previous results and showed that all members of the genus except the type fell into three major clades more closely related to other genera than to the type. Two of those clades had available generic names that were resurrected, and Mann et al. erected a new genus for the third. Additional work on the genus Troglodytes and allies (Rice et al., 1999; Gómez et al., 2005) has shown that: (1) the Timberline Wren (Thryorchilus browni) is a close relative of the genus, (2) the Winter Wren may best be recognized in its own genus, Nannus (although evidence for its exclusion from Troglodytes is not overwhelming); and (3) the Socorro Wren, traditionally placed in Thryomanes, is actually a member of Troglodytes.
FIGURE 1.
Phylogeny of the Certhioidea based on DNA-DNA hybridization (Sibley and Ahlquist 1990), scaled by a measure of genetic divergence (T50H). The quotes around genus Auriparus indicate that its placement is artifactual (possibly a labelling error; see text). Closed circles indicate estimated Bayesian posterior probabilities ≥0.95 for equivalent nodes (taxon sampling differs) in the concatenated gene analyses of Zhao et al. (2016).

Focus of This Study
Although molecular studies have advanced our understanding of certhioid relationships, only two studies have sampled all the relevant deep lineages, and none has densely sampled the most diverse lineage, the wrens. This study addresses both of these gaps. First, I infer a hypothesis of higher-level certhioid relationships including all major lineages, based on a sixgene (one mitochondrial, five nuclear) data set. Second, based on the same data, I infer the first genus-level phylogeny of the wrens (family Troglodytidae). Finally, I review some implications of this phylogeny for the evolution of wrens.
MATERIALS AND METHODS
Taxon and Gene Sampling: This study samples all but one of the currently recognized genera of Certhioidea (Gill and Donsker, 2017), lacking only the polioptilid Ramphocaenus, which is closely related to Microbates (Barker, unpublished data). This includes samples from all four clades of the previously recognized genus “Thryothorus” (true Thryothorus, Pheugopedius, Thryophilus, and Cantorchilus; Mann et al., 2006), both Troglodytes sensu stricto and Nannus (the Winter Wren, which some consider generically distinct; Rice et al., 1999; Gómez et al., 2005), and the monotypic Caribbean endemic genus Ferminia (table 1), which has never before been included in a phylogenetic study. Outgroups for this analysis included members of the Donacobiidae, Cisticolidae, Zosteropidae, Mimidae, Sturnidae, and Turdidae (table 1), as in a previous study of wren relationships (Barker, 2004). In terms of loci, this study includes sequences from one mitochondrial (cytochrome b) and five autosomal nuclear gene regions: RAG1 and RAG2 (recombination activating genes 1 and 2, respectively), FGB (β-fibrinogen, introns 4 and 7), and ZEB1 (zinc finger E-box-binding homeobox 1). All of these loci except ZEB1 (HomoloGene UID #31779, formerly known as TCF8 and δEF1) have been used previously in avian systematics (e.g., Prychitko and Moore, 1997; Barker, 2004; Barker et al., 2004). Similar to RAG1, ZEB1 is a strongly conserved gene with a long exon (though not the sole exon, as in RAG1) that preliminary results indicate is useful in avian phylogenetics (Herreman, 2000).
FIGURE 2.
Summary of well-supported relationships among genera of Troglodytidae based on previous analyses of molecular data (Rice et al., 1999; Barker 2004; Martínez Gómez et al., 2005; Mann et al., 2006).

TABLE 1.
Species and samples included in this study. GenBank accessions for all sequences are listed under each gene.

Continued

Generation of Molecular Data: Genomic DNA was extracted from all samples using a DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). DNA from the sample of Ferminia was subsequently amplified by random priming using the illustra GenomiPhi V2 amplification kit (GE Healthcare, Pittsburgh, PA) to obtain adequate amounts of high molecular weight DNA. All loci were amplified by polymerase chain reaction using previously described primers and cycling conditions (Barker, 2004; Barker et al., 2004; Kimball et al., 2009), although some PCRs for this study were performed using GoTaq G2 Hot Start Master Mix (Promega, Madison, WI). To obtain complete sequences for Ferminia, additional specific primers were created for all loci (appendix 1). For most taxa, ZEB1 was amplified in three fragments using the primer pairs EF31F/EF799R, EF678F/EF1356R, and dEF1x/EF1707R (or in some cases EF_3prime; fig. 3, table 2), using a touchdown cycling profile (five cycles each at 58° C, 56° C, 54° C annealing temperature, followed by 20 at 52° C annealing), and 1 minute extension times. PCR reactions were evaluated by agarose gel electrophoresis, and successful amplifications with appropriately sized products were submitted to Beckman-Coulter Genomics (Danvers, MA) for clean up and sequencing with BigDye Terminator v3.1 on an ABI 3730 automated sequencer, using external and internal primers, as necessary. Individual reads were assembled and edited in Geneious v5.6.7, then exported to text for alignment and subsequent analysis.

Alignment and Phylogenetic Analyses: Sequences for each gene region were aligned using MUSCLE v3.6 (Edgar, 2004) with default parameters, then concatenated for analysis. Certhioid phylogenies were estimated using single gene, concatenation, and species tree analyses in both likelihood and Bayesian frameworks. Likelihood searches as implemented in RAxML v7.0.3 (Stamatakis, 2006) were used to estimate a concatenated gene phylogeny, with a partitioned GTR+G4 (Lanave et al., 1984; Yang, 1994) model allowing proportional branch lengths among partitions. The partitioning scheme was selected using Partitionfinder v1.1.1 (Lanfear et al., 2012), using the greedy search algorithm and BIC as an optimality criterion, and starting with 14 partitions: all codons of each protein coding gene (cytochrome b, RAG1, RAG2, and ZEB1) separately, plus both introns (FGB-I4, FGB-I7). A search for the best tree was made 10 times from random starting points, and nodal support was assessed by 1000 bootstrap replicates (Felsenstein, 1985) using the fast search option. The same partitioning scheme (and model parameterization) was used to estimate relationships for the concatenated data set using Bayesian methods as implemented in MrBayes v3.2.5, using default priors for all parameters except for branch lengths, which were set to an exponential prior with a rate of 100, in order to avoid long branch artifacts identified in initial runs (Marshall, 2010). Bayesian methods were also used to generate gene-specific estimates of phylogeny for comparison with the combined estimate, and to assess among-gene heterogeneity in phylogenetic estimates. For each Bayesian analysis, I performed two MCMC runs each of 2.106 generations, sampling every 100. Adequate (≥200) effective sample size for parameters, parameter convergence, and burn-in were determined using Tracer v1.5, and overall topological and nodal convergence was assessed using functions of the “rwty” package (Lanfear et al., 2016) in R (R Core Team, 2016). A maximum clade credibility tree was calculated from the combined output using TreeAnnotator v1.8.3.
Taxonomic hypotheses of interest were evaluated on the concatenated data by comparing the marginal log-likelihood estimates from unconstrained analyses to values from constrained analyses enforcing monophyly of specific clades using empirical Bayes factors (Kass and Raftery, 1995). Hypotheses evaluated included: (1) monophyly of “Thryothorus” (Thryothorus, Pheugopedius, Thryophilus, and Cantorchilus); (2) monophyly of all former Thryothorus except T. ludovicianus; (3) broad-sense monophyly of Troglodytes (including Nannus, Troglodytes, and Thryorchilus); and (4) monophyly of Troglodytes in the strict sense (Nannus and Troglodytes only). Marginal log-likelihoods were estimated via stepping-stone analyses (Xie et al., 2011) in MrBayes, using 50 steps and α = 0.4, sampling 1.106 generations, with burn-in set to equal the number samples in each step (burninss=-1).
TABLE 2.
Novel primers used in amplification of ZEB1.

In addition to single gene and concatenated analyses, I estimated certhioid phylogeny using Bayesian species-tree methods as implemented in *BEAST v1.8.3 (Heled and Drummond, 2010). Species tree analyses assume free recombination between loci, but no recombination within loci, essentially treating each gene rather than each site as an independent measure of species relationships (Liu et al., 2008; Heled and Drummond, 2010). Since two pairs of the six loci included in this study—FGB-I4/FGB-I7 and RAG1/RAG2—are closely physically linked (separated by 1251 and 8042 bases respectively in Taeniopygia; GenBank assembly 3.2.4, Annotation release 101), I treated these pairs as single loci in terms of topology (but not in substitution model), yielding effectively four independent loci for the purposes of species tree analysis. Ploidy was set as mitochondrial for cytochrome b, and autosomal for the remaining loci, based on assumed synteny with Taeniopygia. I set a Yule prior on the species tree topology, used a piecewise constant multispecies coalescent model, and assumed an uncorrelated log-normally distributed model of lineage-specific rate variation. Priors on gene-specific rates were set as exponential distributions with means of 0.1. I performed two MCMC runs each of 1.108 generations, sampling every 5000. Run outputs were analyzed as for the concatenated and single gene analyses reported above.
It was previously shown that RAG1 and RAG2 sequences of Sitta deviate strongly from stationarity, showing an excess of GC nucleotides at third-codon positions in comparison with other passerines (Barker et al., 2002; Barker et al., 2004). I assessed base composition variation at all loci using taxon-specific goodness-of-fit tests (Gruber et al., 2007). Sequence subsets for individual taxa showing significant departures from the overall mean were recoded as appropriate (e.g., AGY coding for mitochondrial DNA third positions, or RY coding for nuclear gene third positions, Phillips and Penny, 2003; Gibson et al., 2005), and potentially affected analyses rerun.
Integrated Analysis with Published Data: As noted above, Zhao et al. (2016) recently published an analysis of higher-level certhioid phylogeny. This study used data largely independent of those reported here; consequently, it is possible that integrated analysis of the two data sets could yield better support for basal relationships than achieved in either. To evaluate this, I constructed the largest complete matrix possible at the generic level. This yielded a data set of six certhioid taxa (Sitta, Tichodroma, Certhia, Salpornis, Polioptila, and Nannus) and two outgroups (a chimeric “cisticolid” including sequences of Prinia and Cisticola; and Sturnus). These taxa were sampled for a total of 10 gene regions (the six described above, plus GAPDHI11, LDH-I3, MB-I2, ODC-I6/7), for a total of 11,883 aligned base pairs. Alignments for the Zhao et al. data were performed as described above for the new data reported here. The concatenated data were analyzed using partitioned maximum likelihood and Bayesian methods as described above. In addition, the data for completely sampled loci were analyzed using species tree methods as described above, recognizing six independent gene regions: CYTB, FGB (I4+I7), MB-I2, ODC- I6/7, RAG1+RAG2, and ZEB1.
TABLE 3.
Characteristics of gene regions sampled in this study. Shown are the locus names, linkage (based on the Taeniopygia genome), alignment length, numbers of variable and informative characters, and results from single gene analyses using MrBayes v3.2.5 (tree lengths, number of partitions receiving ≥0.95 estimated posterior probability, and mean parameter values under a GTR+I+G4 parameterization.

RESULTS
Data Characteristics and Single Gene Analyses: I obtained sequence for all taxa from all loci, though a handful of taxa were incomplete for some loci, including Salpornis missing the 5′ half of FGB-I4, three species missing 83–133 bases from FGB-I7, and Nannus missing 323 bases from RAG1. All sequences have been submitted to GenBank (see table 1 for accessions), and the concatenated alignment is available at TreeBase (study ID 21870). The alignments obtained from these data ranged in size from 633 (FGB-I4) to 2876 (RAG1) bases in length, with 119 (FGB-I4) to 401 (CYTB) phylogenetically informative sites (table 3). In terms of percentages, ZEB1 yielded the fewest informative sites per sequenced base (4%), the other nuclear loci were approximately equivalent to one another (10%–19%), and CYTB yielded the most (38%). Bayesian phylogenetic analyses of these data sets under a uniform GTR+I+G4 parameterization revealed significant heterogeneity in substitution dynamics among these loci (table 3), as expected given their location in differing genomes (mitochondrial versus nuclear) and variation in coding status (introns versus exons). In particular, based on estimated tree lengths, mitochondrial CYTB evolved at approximately 5x (ranging from 2.8–9.1) the rate of the nuclear genes, and showed much stronger base compositional bias (table 3). In addition to variation among genes, some loci showed variation in base composition among taxa (online supplementary table 1: https://doi.org/10.5531/sd.sp.28). Base frequencies at third positions of Sitta RAG1 deviated significantly from other taxa in the sample (with RAG2 showing a similar, nonsignificant trend), as did third positions of Dumetella CYTB. In the case of CYTB third positions, a number of other taxa showed substantial (but nonsignificant) deviations, primarily in the relative proportion of cytosine and thymine bases, as noted previously for mitochondrial genomes (Gibson et al., 2005; Powell et al., 2013). To assess the impact of these deviations, phylogenetic analyses of data from these genes and taxa were repeated with RY (RAG1) and AGY (CYTB) coding.
Concatenated Analyses: Model fitting for partitions of the concatenated-sequence data set identified a strategy with seven partitions: (1) CYTB 3rd positions, (2) CYTB 1st positions, (3) CYTB 2nd+ZEB1 3rd positions, (4) FGB-I4+FGB-I7+RAG2 3rd positions, (5) RAG1 3rd positions+RAG2 1st and 2nd positions, (6) RAG1 1st and 2nd positions, and (7) ZEB1 1st and 2nd positions. Both maximum-likelihood and Bayesian analyses of the data with this model, recoding non-stationary sites as identified above, yielded a single tree (fig. 4). In this instance, data recoding appeared to have little impact on the inferred tree, and the best tree from the original data was nearly identical (not shown), differing only in recovery of Salpornis and Certhia as sister taxa, and bootstrap values were nearly indistinguishable (not shown). Notably, 17 of 31 bipartitions in concatenated analysis received substantial support (defined as ≥0.90 posterior probability) from at least three individual genes (fig. 5). Concatenated support for this tree was generally good, with 18/31 bipartitions receiving ≥75% bootstrap values, and 21/31 receiving ≥0.95 estimated posterior probabilities (plotted on the concatenated tree estimate in fig. 4). Resolution was best for outgroup relationships and relationships among the wrens and allies. In particular, these data supported: monophyly of the Certhioidea relative to the outgroups included here, monophyly of the New World certhioids (Troglodytidae + Polioptilidae) and of the two families in the clade; and many intergeneric relationships within the Troglodytidae. By contrast, basal relationships among certhioids were not well resolved by these data, with the strongest support being for separation of Sitta from all other taxa in only 59% of bootstrap replicates of the recoded data (fig. 4).
FIGURE 4.
Best estimate of certhioid phylogeny, based on concatenated analysis of 8517 bases from five gene regions (cytb, FGB-I4, FGB-I7, RAG1, RAG2, and ZEB1). Shown is the maximum-likelihood tree from a partitioned RAxML analysis, with nonstationary partitions AGY-recoded (see text). Bootstrap support values (from 1000 fast bootstrap replicates; left) and estimated posterior probabilities from partitioned, concatenated Bayesian analysis (right) are shown below each branch. Node numbers correspond to barplots in figure 5.

Among-gene Conflict and Species Tree Analyses: Single-locus analyses indicated several instances of significant conflict over phylogenetic relationships (fig. 5). In four cases (nodes 6, 18, 19, and 22), one (but never more than one) gene region showed strong (≥0.90 posterior probability) conflict with one or more genes supporting that relationship. In an additional four cases (nodes 3, 21, 29, and 30) single genes strongly conflicted with the consensus topology, although no other individual genes strongly supported it. Presumably due to this conflict, only two of these eight nodes (6 and 19) were consistently supported in both likelihood and Bayesian concatenated analyses (figs. 4, 6). Species tree analyses with *BEAST accounting for this amonglocus heterogeneity in history yielded a maximum clade credibility tree (fig. 6) congruent with concatenation analyses, with two exceptions. These exceptions were placement of Certhia and Salpornis as sister taxa, and placement of Salpinctes as sister to Catherpes+Hylorchilus: neither of these relationships received strong support from either the concatenated or species tree analyses. As expected given the evidential shift from sites to loci, support for certhioid relationships in species tree analyses was generally lower than for concatenated analyses, with 25/31 bipartitions in the species tree receiving higher posterior probabilities in concatenation. However, six bipartitions actually had higher support in species tree analysis, most notably including: monophyly of a clade of wrens excluding Catherpes, Hylorchilus, Microcerculus, Salpinctes, and Odontorchilus, and a sister group relationship between Henicorhina and Cyphorhinus.
FIGURE 5.
Evaluation of support and conflict for individual nodes recovered in concatenated analysis of five gene regions. For each node (see fig. 4 for definitions), a barplot indicates the strength of support for that node found in independent analyses of each gene region using MrBayes with a GTR+I+G4 parameterization as dark bars above the abscissa, as well as the strength of support against that node (defined as the highest support for all conflicting nodes recovered; light bars below the abscissa). The estimated posterior probabilities from concatenated analysis are shown after each node number.

FIGURE 6.
Best estimate of certhioid phylogeny, based on species tree analysis of five gene regions (cytb, FGB-I4, FGB-I7, RAG1, RAG2, and ZEB1). Shown is the maximum clade credibility tree from analysis with *BEAST v1.8.3, coding the data as four “genes” based on close linkage of several gene regions. Nodes with estimated posterior probabilities ≥0.95 from partitioned, concatenated Bayesian analysis are indicated by black circles, and those also receiving support at the same level in species tree analyses are indicated by gray circles with black outline.

Phylogenetic Hypothesis Tests: Specific a priori hypothesis testing was performed for the concatenated data (table 4). All alternative hypotheses except one were very strongly rejected (Kass and Raftery, 1995) by empirical Bayes factor comparisons with the unconstrained analysis. The exception was monophyly of the genus Troglodytes in the broad sense (i.e., including Nannus, Thryorchilus, and Troglodytes), which had a marginal likelihood essentially indistinguishable from the unconstrained analysis.
TABLE 4.
Results of constrained Bayesian analyses of the concatenated data. Shown are the stepping-stone (SS) estimates (Xie et al., 2011) of marginal likelihoods from an unconstrained analysis and analyses with several a priori phylogenetic constraints, as well as empirical Bayes factors (twice the natural log of K, 2lnK) comparing constrained to unconstrained likelihoods.

Integrated Analysis with Published Data: Analysis of the data reported here in conjunction with the previously published data of Zhao et al. (2016) yielded a slightly more resolved picture of basal certhioid relationships (fig. 7). In particular, concatenated analyses strongly supported a sister-group relationship between Salpornis and Certhia, although this relationship only had a posterior probability of 0.28 in species tree analysis, and cytochrome b strongly conflicted with this relationship (fig. 8). As found in analyses of the broader taxon sample with fewer loci (see above), monophyly of Certhioidea was strongly supported (albeit with only two outgroups), as was a sister-group relationship between the wrens (Troglodytidae) and gnatcatchers (Polioptilidae). Relationships among other certhioid lineages (Sitta and Tichodroma) remained unresolved.
DISCUSSION
Basal Certhioid Relationships
The two previous studies with adequate higher-level sampling of certhioids (Sibley and Ahlquist, 1990; Zhao et al., 2016) were completely congruent in their estimate of relationships in the group (fig. 1). The current study does not significantly contradict previous results, and does little in the way of corroboration. In particular, concatenated analysis of the data reported here fails to recover the previously reported sister-group relationships between Tichodroma and Sitta and between Salpornis and Certhia, although the latter relationship was recovered in species tree analyses (with poor support). Perhaps the most notable contribution to resolving certhioid relationships here is an increase in support for the sister-group relationship between Salpornis+Certhia and the New World wren/gnatcatcher clade. Although Zhao et al. (2016) recovered this relationship in concatenated analysis, it only received a posterior probability of 0.82, and it was not found in the species tree. In combined analyses of most of Zhao et al.'s data and the new data reported here (fig. 7), this relationship received a posterior probability of 0.99 in concatenated analysis, and was also recovered in the species tree analysis, albeit with an estimated posterior of 0.40. It is likely that phylogenomic approaches (e.g., ultraconserved elements; McCormack et al., 2013) will be necessary to resolve basal relationships of this group with certainty.
FIGURE 7.
Best estimate of certhioid phylogeny, based on concatenated analyses of 10 gene regions (cytb, FGB-I4, FGB-I7, GAPDH-I11, LDH-I3, MB-I2, ODC-I6/7, RAG1, RAG2, and ZEB1). Shown is the maximum clade credibility tree from partitioned analysis with MrBayes. Nodes with estimated posterior probabilities ≥0.95 from partitioned, concatenated Bayesian analysis are indicated by black circles, and those also receiving support at the same level in species tree analyses with *BEAST (coding the data as six “genes” based on close linkage of several gene regions) are indicated by gray circles with black outline. In addition, bootstrap support values (1000 fast bootstrap replicates) from partitioned concatenated analysis with RAxML are shown below each branch. Node numbers in circles correspond to barplots in figure 8.

Basal Relationships of Troglodytidae
Both the gene and taxon sampling of this study are the best to date for addressing higherlevel relationships of wrens and gnatcatchers. As expected based on previous results (reviewed above), wrens and gnatcatchers form a well-supported clade within Certhioidea. Within the wrens, there is strong support for a basal split between a small clade of wrens with terrestrial habits (Microcerculus, Salpinctes, Catherpes, and Hylorchilus; a total of eight species, termed here the geophilous wrens) and all other wrens. Previous studies (Barker, 2004; Mann et al., 2006) were ambiguous regarding the rooting of the wren tree, with one indel in FGB-I4 (Barker, 2004: indel 6) pointing to a root at Salpinctes, a member of the geophilous wren clade. Based on analysis of nucleotide variation in the genes sampled here, it is apparent that that indel was either homoplastic or a misalignment. Reexamination of the alignment shows that the indel involves a simple 11 base pair tandem repeat that has diverged by 1 base pair in Salpinctes, driving its alignment against the second repeat unit that is identical to Salpinctes in an outgroup, rather than alignment with other wrens.
Perhaps the only outstanding question regarding basal relationships of wrens is the placement of the South American endemic genus Odontorchilus. This involves one of only four cases of hard incongruence among genes in this data set (fig. 5: node 18). Two genes (FGB-I4 and RAG2) strongly support placement of the genus outside the main radiation of nongeophilous wrens, whereas one gene (FGB-I7) strongly supports its placement as sister to one of the two main clades in this radiation. A third gene (RAG1) is congruent with RAG2, but with support just below 0.9 posterior probability. Odontorchilus wrens have long been recognized as distinct, in particular due to their toothed bill (for which they are named), their preferred foraging stratum in the canopy, and their simple trilled songs; consequently, no there is no clear a priori expectation for their placement anywhere within wrens. However, it is interesting to find a South American lineage placed so deeply within the family (see below).
Relationships of Nongeophilous Wrens
The nongeophilous wrens apart from Odontorchilus are divided into two well-supported clades (fig. 4: A, B) of nearly the same number of genera (seven and eight, respectively), and species (42 and 36, respectively). Clade A comprises subtropical and tropical species best known for their singing ability and nearly ubiquitous habit of performing vocal duets (Mann et al., 2009). Clade B comprises genera with both tropical and temperate distributions, including three small genera (two monotypic) with only temperate species. Many more species in this clade do not perform vocal duets, and species in two genera nest in tree cavities, a behavior otherwise only known from the genus Microcerculus, which nest in tunnels in the soil.
Clade A includes three genera formerly subsumed in the genus Thryothorus, until molecular data strongly supported placement of the type of that genus (the Carolina Wren T. ludovicianus) as sister to Thryomanes, to the exclusion of all other species that had been placed in it (Barker, 2004; Mann et al., 2006). Mann et al. (2006) recognized three clades of former “Thryothorus” wrens as genera (Pheugopedius, Thryophilus, and Cantorchilus), giving a new name to the third. This treatment of these taxa is strongly corroborated by the current study. In particular, Bayes factor comparisons strongly reject association of Thryothorus ludovicianus with the other former members of the genus, as expected given previous analyses. In addition, Bayes factors strongly reject a monophyletic origin of the three genera in which former Thryothorus species are now placed, suggesting that these species cannot be subsumed under the oldest generic name (Pheugopedius) for the sake of simplicity alone. Relationships within this clade are generally strongly supported in concatenated Bayesian but not in concatenated-likelihood or species tree analyses (figs. 4, 6). Notably, the genus Pheugopedius is strongly supported (except in species tree analysis) as sister to all other genera in the group. In concatenated Bayesian analyses, Cantorchilus is strongly supported as sister to Cinnycerthia, and Thryophilus as sister to Uropsila, explaining why monophyly of former Thryothorus species is strongly contradicted by these data. Relationships of Henicorhina and Cyphorhinus are less clear, with maximum-likelihood analysis (fig. 4) favoring them as sequential sister taxa to Uropsila+Thryophilus, and species tree analysis (fig. 6) placing them as sister taxa (neither relationship with appreciable support).
Relationships among Clade B wrens were better resolved than those in Clade A. Four of six intraclade relationships were strongly supported by both concatenated and species tree analyses (figs. 4, 6). These well-supported relationships included the previously recovered sister-group relationship between the monotypic genera Thryothorus and Thryomanes, and their sister-group relationship to the diverse (both phenotypically and in species numbers) genus Campylorhynchus. The remaining four genera of Clade B formed a well-supported sister clade to these three. The only strongly supported relationship among these four genera placed the genus Troglodytes (a diverse, cosmopolitan, but relatively morphologically uniform group) sister to Thryorchilus (a distinct monotypic genus of the Central American highlands). It is possible that this close relationship is actually due to Thryorchilus falling within Troglodytes, although the one study with broader sampling of the latter suggests otherwise (Gómez et al., 2005). The remaining ambiguities lie in the relative placement of the genera Nannus, Cistothorus, and Ferminia.
FIGURE 8.
Evaluation of support and conflict for individual nodes recovered in concatenated analysis of 10 gene regions (results from two incompletely-sampled loci not shown). Support and conflict are shown as in figure 5 (see fig. 7 for corresponding node definitions).

The genus Nannus is the only lineage of the wren and gnatcatcher clade with species in the Old World, comprising at least three species Holarctic in distribution (Drovetski et al., 2004). Until recently, these birds were classified as a single species in the genus Troglodytes: the Winter Wren, T. troglodytes. Mitochondrial studies have consistently pointed toward a distant relationship of Nannus species to core Troglodytes, with Thryorchilus and possibly Cistothorus intervening between the two (Rice et al., 1999; Gómez et al., 2005). In the current study, both Cistothorus and the genus Ferminia (never before included in a molecular phylogeny) separate Nannus from Troglodytes, though neither relationship showed substantial support (figs. 4, 6). Bayes factor comparison of these results to an analysis with Troglodytes monophyly constrained strongly favored the former (table 4). However, the marginal likelihood of an analysis with Nannus, Thryorchilus, and Troglodytes constrained as monophyletic was indistinguishable from the unconstrained analysis (table 4), indicating that the strongest signal is for monophyly of Troglodytes+Thryorchilus. Thus, these data would not contradict a classification that subsumed all three genera (Nannus, Thryorchilus, and Troglodytes) within Troglodytes, as previously done by its describer (Bangs, 1902), and some subsequent taxonomies (e.g., Paynter and Vaurie, 1960). Unless subsequent data strongly separate Nannus from Troglodytes+Thryorchilus, this would seem appropriate, despite the distinctiveness of Thryorchilus.
The remaining two lineages in this unresolved region of the wren tree are Cistothorus and Ferminia. The former genus comprises at least five species distributed from northern North America through the cape region of South America (including the Islas Malvinas). All Cistothorus species are associated with grasslands and marsh habitats, and one widespread species (C. platensis) is found in grasslands from Central America nearly to Cape Horn (though this species is in need of splitting; Robbins and Nyári, 2014). The genus Ferminia is monotypic, with the single species F. cerverai endemic to Cuba, where it is restricted to the Zapata swamp of southern Matanzas province (Garrido and Kirkconnell, 2000). The species exhibits some interesting parallels with Cistothorus, including living in a marsh habitat, construction of woven domed nests on grasses or emergent vegetation (Martínez and Martínez, 1991; Lianes Sosa and Mancina, 2002; Forneris and Martínez, 2003), and vocal similarities. Based on recordings and videos (Internet Bird Collection, http://www.hbw.com/ibc/species/zapata-wren-ferminia-cerverai; Xeno-canto, http://www.xeno-canto.org/species/Ferminia-cerverai; both accessed September 2017), male Ferminia have vocal repertoires (e.g., see Xeno-canto catalog XC256894), as in many wren species (e.g., Kroodsma, 1975; Kroodsma and Verner, 1978; Molles and Vehrencamp, 1999; Logue, 2006; Bradley and Mennill, 2009), and may engage in matched countersinging as seen in some Cistothorus (Kroodsma and Verner, 1978). At least some Ferminia songs include a series of repeated low-frequency syllables most closely matched among wrens, based on my extensive listening to wren vocalizations both in the field and in recordings, by songs of the marsh wren C. palustris (e.g., fig. 9). In terms of plumage, it is perhaps most similar to Troglodytes, with dull brownish-dun underparts and richer brown strongly barred upperparts (first figured by Brooks in Barbour, 1928, or see Forneris and Martínez, 2003, for a photograph), a similarity noted but dismissed by Barbour in his description of the species (Barbour, 1926). Consequently, it is perhaps unsurprising to find this species in an indeterminate position relative to these two genera. Regardless of how these relationships are ultimately resolved, this mosaic pattern of phenotypic similarities and closely spaced divergences suggests relatively rapid diversification at the base of this group.
FIGURE 9.
Comparison of song characteristics between Ferminia cerverai and Cistothorus palustris. Shown is the spectrogram of a single Ferminia song (seconds 1.93–4.28 of Xeno-Canto catalog number XC256894, recorded by Hans Matheve and reproduced with permission), compared to the spectrogram of a single C. palustris song (seconds 1.77–2.92 of Xeno-Canto catalog number XC386079, recorded by the author) with a similar repeated syllable. Both spectrograms were generated using the “spectro” function of the R package “Seewave” (Sueur et al., 2008; the C. palustris recording was also filtered from 0–1.1 kHz to remove low-frequency background noise).

Biogeography and Diversification of Certhioidea
Although not well resolved phylogenetically, the earliest divergences within Certhioidea are among Old World or ancestrally Old World lineages (fig. 10). Both Tichodroma and Salpornis are exclusively Old World, and both Certhia (Tietze et al., 2006) and Sitta (Pasquet et al., 2014) clearly have Old World origins, despite each lineage having invaded the New World (once in Certhia and three times in Sitta). Thus, certhioids are undoubtedly Old World (possibly north temperate) in origin, consistent with an Old World origin for the entire certhioidmuscicapoid clade and indeed for oscines as a whole (Barker et al., 2004; Moyle et al., 2016). While dispersal of Certhia and Sitta into the New World has not resulted in substantial diversification (a total of six species by current taxonomy, but possibly as many as 10; Manthey et al., 2011; Walstrom et al., 2012), another certhioid lineage—the ancestor of the wrens and gnatcatchers—dispersed into the New World and diversified both in species number (a total of 106 species; Gill and Donsker, 2017) and in ecologically related phenotypic traits such as body size, limb proportions, and beak shape.
FIGURE 10.
Biogeography of certhioid lineages. Shown is the species tree estimate for certhioids, scaled to absolute time using an estimated age of 16.1 Ma for the divergence of Certhia and Troglodytes (Moyle et al., 2016). The continental distribution of each lineage is shown to the right, with black fill indicating presence, and light fill absence.

Based on a secondary point calibration (Moyle et al., 2016), the New World Certhioidea (NWC) most likely dispersed from the Old World between 16.1 (stem age) and 11.8 million (crown age) years ago, the mid-Miocene (fig. 10; this represents a minimum range, since no uncertainty was included in scaling of this tree). If this range of estimated dispersal times is accurate, it suggests a north Pacific route into the New World via Beringia, rather than a North Atlantic or Antarctic route (Sanmartín et al., 2001). No extant member of the NWC is a strong flier, and long-distance dispersal directly into South America seems unlikely. Consequently, it is likely that the NWC diversified in North America prior to independent invasion of South America by multiple lineages, as has been previously suggested for wrens (Mayr, 1946), and as seems the case for emberizoids (Barker et al., 2015). Within wrens, this is corroborated by the primarily North American distribution of the geophilous wrens (half of the first split in the Troglodytidae); however, the relatively deep placement of the South American Odontorchilus suggests early dispersal into South America (or possibly extinction of this lineage from the north; fig. 10). By contrast, all three genera of Polioptilidae are found in both North and South America: if possible, resolution of the ancestral area of this clade will require species-level as well as (most likely) extensive intraspecific sampling.
Assuming a northern origin, a minimum of six dispersal events into South America are required to explain the extant distribution of wrens, including dispersals of Microcerculus, Odontorchilus, Clade A, Campylorhynchus, Cistothorus, and Troglodytes (fig. 10). In some cases, extant distributions would most likely require additional dispersals south or back dispersals from the south to the north: current taxon sampling does not merit a formal analysis. Nevertheless, it is clear that the NWC has had a long history in both North and South America. In particular, the origin of Clade A by dispersal into South America would imply presence of wrens in the Pliocene or earlier, supporting either overwater dispersal of this lineage (not unreasonable given the presence of Troglodytes wrens on islands as remote as Isla Clarión and Isla Socorro, although it is worth noting that species in this genus are migratory), or early closure of the isthmus, as has recently been hypothesized (Bacon et al., 2015; Montes et al., 2015; but see O'Dea et al., 2016). Complete taxon sampling of this lineage may clarify this history, in particular the timing and directional bias of dispersal events and subsequent diversification rates (e.g., Barker et al., 2013; Barker et al., 2015).
ACKNOWLEDGMENTS
I thank the curators and collections managers at the American Museum of Natural History, the Biodiversity Institute (University of Kansas), the Burke Museum (University of Washington), the Museo de Zoología “Alfonso L. Herrera” (Universidad Nacional Autonoma de México), the Field Museum, and LSU Museum of Zoology for loans of material in their care. This paper benefitted from comments by Shanta Hejmadi, Tyler Imfeld, and Michael Wells. This work was supported by a grant from the NSF Biological Sciences program, DEB-1541312.