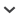Rapidly evolving introns of low-copy nuclear markers have the potential to generate robust species-level phylogenetic hypotheses (Sang, 2002). With current high-throughput sequencing technologies, genomic and transcriptomic data sets are becoming available to use for the development of informative markers for phylogenetic utility. In the most fortunate of cases, the available genome or transcriptome is within the targeted lineage of interest or is closely related (Curto et al., 2012; Granados Mendoza et al., 2015; Stockenhuber et al., 2015). In other cases, authors generate transcriptome data of members of the targeted group (Tonnabel et al., 2014; Stockenhuber et al., 2015). Combining these data, authors aim to target low-copy nuclear markers for phylogenetic utility. Although this strategy is very promising, whole genomes are not typically available for nonmodel systems, and generating transcriptome data de novo is expensive and may be unnecessary given existing data (e.g., 1000 Plants [1KP] project; www.onekp.com).
Here, we present a bioinformatic pipeline that leverages a publicly available genome and published transcriptome reads to identify conserved regions in single-copy nuclear loci and to design primers for amplification of associated introns. Benefits of this pipeline include (1) reduced cost by not generating sequence data de novo and (2) targeting nuclear introns, which are expected to have high sequence variation even among closely related species. This pipeline can be powerful in cases where published transcriptomes are phylogenetically closer to the targeted lineage than the available genome, and in cases where researchers are interested in single-copy nuclear introns for phylogenetic resolution. It is useful for researchers interested in using small-scale sequencing efforts (i.e., Sanger sequencing) to identify relatively few (1–20) informative nuclear loci that can be amplified by PCR, but could be scaled up to include larger sets of loci by relaxing parameters and/or reducing the number of filtering steps in the pipeline. The final set of loci can be used to design baits for Hyb-Seq next-generation sequencing (Weitemier et al., 2014), homemade in-solution capture (Peñalba et al., 2014), or microfluidic PCR primers (Uribe-Convers et al., 2016).
We demonstrate the utility of this bioinformatic pipeline to design primers to amplify single-copy nuclear introns in the tribe Paullinieae (Sapindaceae), a Neotropical lineage of ∼475 liana species (Acevedo-Rodríguez et al., 2017). A previous phylogenetic analysis of the Paullinieae tribe was strictly morphological (Acevedo-Rodríguez, 1993) and only at the generic level. Most recently, Acevedo-Rodríguez et al. (2017) aimed to resolve generic- and species-level relationships in Paullinieae using ITS and the trnL intron. Although important tribal relationships were resolved, these two markers resulted in a polytomy of Serjania Mill., Paullinia L., Urvillea Kunth, and Cardiospermum L. Thus, more phylogenetically informative molecular markers are needed to improve the resolution of generic- and species-level relationships in Paullinieae. Here, we describe a bioinformatic pipeline that leverages publicly available genomic data from distantly related lineages (within the order and within the family but distant to the tribe of interest) to successfully design primers for a specific tribe. We demonstrate the pipeline using the annotated Citrus sinensis (L.) Osbeck (Rutaceae: Sapindales) genome with two sets of transcriptome reads, Dimocarpus longan Lour. and Litchi chinensis Sonn., that are within Sapindaceae but outside of the Paullinieae tribe of interest. The estimated pairwise divergence time between the transcriptomes and genome is 94 mya ( www.timetree.org). By using two sets of transcriptome reads from species in Sapindaceae, we were able to find conserved regions from which primers could be designed for amplification of single-copy nuclear introns.
METHODS AND RESULTS
Finding single-copy nuclear markers—To generate nuclear intron markers, the annotated genome of C. sinensis (Rutaceae) provided the coding sequences, intron positions, and estimated intron size while the transcriptomes of two more closely related Sapindaceae species, D. longan and L. chinensis, provided the best estimate of the gene sequence from which primers could be designed for the target lineage. By using this combination of available data, we hoped to identify single-copy gene regions to avoid amplification of unidentified paralogs, and to design primers that would have a high likelihood of amplification success across the Paullinieae (Sapindaceae).
First, the genome is processed: genome coding sequences (CDS) were downloaded and filtered for single isoform mRNA strands for ease of processing, and genes with introns of 500–1100 bp were selected so they would be easily amplified by traditional PCR and still contain a sufficient number of characters to have phylogenetic utility. The number of base pairs could be changed to include greater or fewer regions as desired. Second, the transcriptome reads are cleaned to remove adapters, low-complexity sequences, contamination, and PCR duplicates (Singhal, 2013). Third, a series of steps are applied to use the genome data to obtain homologous sequences from the more closely related transcriptome without generating a transcriptome assembly de novo as in Sass et al. (2016): (1) Cleaned transcriptome reads are aligned to the filtered genome coding sequences using NovoAlign version 3.01 (Novocraft Technologies, Petaling Jaya, Selangor, Malaysia; http://novocraft.com) with -t 480, a lenient value that allows highly divergent sequences to map. (2) Single-nucleotide polymorphisms (SNPs) are called using SAMtools version 0.1.19 (Li et al., 2009), and new consensus sequences are generated based on the SNPs called. (3) Transcriptome reads are aligned to the new consensus sequences created from the first alignment using NovoAlign -t 90 (a more stringent value). (4) SNPs are called and the final consensus sequences are created to serve as a pseudoreference for primer design. The iterative alignment and SNP calling enables more distantly related transcriptome reads to align to the genome. The following filters are applied to all pseudoreference sequences (Fig. 1) in this order: (1) Retain sequences that only BLAST to self with default settings (i.e., exclusively BLAST to the Citrus CDS from which the pseudoreference was generated). (2) Remove sequences that BLAST to plastid, chloroplast, ribosomal, transposon, or mitochondrial loci using MegaBLAST and the National Center for Biotechnology Information (NCBI) database (Organism: Spermatophyta; http://www.ncbi.nlm.nih.gov/). (3) Retain only genes with at least 20× average read coverage (Nielsen et al., 2011). (4) Remove sequences with hits to RepeatMasker ( http://repeatmasker.org/) (i.e., interspersed repeats and low-complexity DNA sequences). After the above filters are applied, these sequences fit the criteria of single-copy nuclear genes containing introns between 500–1100 bp.
Primer design—To verify that primer regions were conserved within the breadth of phylogenetic interest, a second transcriptome within the family of interest was aligned to the pseudoreference. This step increases the chances that the primers will be conserved and utilizable across the breadth of the lineage of interest. This step could be eliminated if a second transcriptome is not available; however, further primer testing would likely be necessary. Litchi chinensis (Sapindaceae) transcriptome reads were aligned to the Dimocarpus pseudoreferences using the Map to Reference tool in Geneious version 8.0.4 (Biomatters Ltd., Auckland, New Zealand; Fig. 1; Appendix 1). Primers were then designed at a conserved coding sequence flanking intron positions using Primer3 version 0.4.0 (Koressaar and Remm, 2007; Untergasser et al., 2012).
Fig. 1.
Bioinformatic pipeline to target single-copy nuclear intron markers. CDS = coding sequences; NCBI = National Center for Biotechnology Information; SNP = single-nucleotide polymorphism.

DNA extraction and primer testing—Several taxa were chosen that represent the phylogenetic breadth of the target lineage. These taxa were used to test the PCR primers with the aim that the primers would work in all samples in the tribe. Initial PCRs used a temperature gradient for the annealing step with a single taxon to determine the optimal annealing temperature for each primer pair. Once the annealing temperature and number of cycles were determined, the optimal PCR conditions were applied to all samples. Only primer pairs that yielded a single band across all samples were sequenced. The sequences were assembled, edited, and aligned and pairwise identity and parsimony informative scores were generated for each marker to determine phylogenetic utility.
Case study: Targeting single-copy nuclear introns of Paullinieae (Sapindaceae)—The ‘Citrus sinensis CDS’ (46,147 sequences; C. sinensis genome version 1.0) and genome annotation files were downloaded from the Citrus Genome Database ( www.citrusgenomedb.org [accessed 22 August 2015]; Wu et al., 2014). Sequences in this CDS file are mature mRNA strands void of introns and untranslated regions. Of these 46,147 mature mRNAs, 18,384 were single isoform mRNAs and, of those, 2159 had introns of 500–1100 bp. The D. longan transcriptome paired-end reads (SRR412534) were downloaded from the NCBI database using the NCBI SRA Toolkit version 2.4.5-2 and cleaned to remove adapters, low-complexity sequences, contamination, and PCR duplicates (Singhal, 2013). Of the 64,876,258 D. longan transcriptome reads, 39,701,810 remained after cleaning. Of the cleaned reads, 573,149 aligned to the 2159 genes from C. sinensis in the final alignment. In transforming the C. sinensis reference to a Dimocarpus pseudoreference, 103,088 SNP positions were changed. After removing low-coverage genes, 1547 pseudoreference sequences remained. The following filters were applied (Fig. 1) in this order: (1) retain sequences that only BLAST to self (by BLAST to both the entire Citrus CDS and against all pseudoreferences) (315 removed); (2) remove sequences that BLAST to plastid, chloroplast, ribosomal, transposon, or mitochondrial loci using MegaBLAST and the NCBI database (Organism: Spermatophyta) and compiled Sapindales plastomes (downloaded from https://www.ncbi.nlm.nih.gov/genbank) (150 removed); (3) retain only genes with at least 20× average read coverage (793 removed); (4) remove sequences with hits to RepeatMasker ( http://repeatmasker.org/) using default settings (i.e., interspersed repeats and low-complexity DNA sequences) (243 removed). After applying the above filters, a total of 46 sequences were isolated that fit the criteria of single-copy, single isoform nuclear genes containing introns between 500–1100 bp. To verify that primer design regions were conserved within the family, a second set of Sapindaceae transcriptome reads, from Litchi chinensis (NCBI: SRX258094; Li et al., 2013), was aligned to the pseudoreference using the Map to Reference tool in Geneious version 8.0.4 at low sensitivity and for two iterations (Fig. 1). Geneious version 8.04 was used at this step because the number of remaining loci made computation on a desktop computer possible and visualization manageable. Of the 53,437,444 L. chinensis reads, 88,779 mapped to the 46 genes of interest. Primers were designed using Primer3 version 0.4.0 (Koressaar and Remm, 2007; Untergasser et al., 2012) by randomly selecting 21 conserved Sapindaceae CDS–CDS boundaries within the Geneious mapped alignment. The L. chinensis reads were not filtered for our purposes, because if primers designed in conserved regions from this second alignment resulted in amplification of multiple PCR products, marker development of these loci was not pursued. However, according to best practices all transcriptome reads should be cleaned prior to mapping. See Appendix 1 for step-by-step instructions.
Taxon sampling, DNA extraction, and primer testing—DNA was extracted from silica-dried leaf material from 12 Sapindaceae samples representing nine species (Appendix 2) using the cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987) with minor modifications. Eleven ingroup samples (Serjania paucidentata DC., S. pyramidata Radlk., S. atrolineata C. Wright, S. mexicana (L.) Willd., Paullinia turbacensis Kunth [3 individuals], P. bracteosa Radlk. [2 individuals], Paullinia sp., and P. glomerulosa Radlk.) and one outgroup species (Allophylus psilospermus Radlk.) were used. After testing annealing temperatures over a temperature gradient (62–43°C) for each primer pair in a representative species, optimized PCR conditions were applied to all samples. Loci were amplified using Phire Hot Start II DNA Polymerase (Thermo Fisher Scientific, Pittsburgh, Pennsylvania, USA) with a 5-min initial denaturing step at 98°C; loci-specific cycles of 5 s at 98°C, 5 s at loci-specific annealing temperature, and 20 s at 72°C; and a final 1-min 72°C extension. Optimal annealing temperatures are reported in Table 1. Only primer pairs that yielded a single band across all samples were sequenced. Cycle sequencing was performed using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Waltham, Massachusetts, USA). Sanger sequencing was done at the UC Berkeley Evolutionary Genetics Laboratory on an Applied Biosystems 3730x1 DNA analyzer. Reads were assembled, edited in Geneious version 8.0.4, and aligned using MAFFT version 7.271 (Katoh and Standley, 2013). The Paullinieae introns were consistently smaller than expected based on the C. sinensis intron sizes. Pairwise identity was calculated in Geneious version 8.0.4, and parsimony informative sites were calculated in PAUP* 4.0b10 (Swofford, 2002) (Table 2). Of the 21 primer sets tried, seven resulted in multiple products, two failed to amplify, and 12 resulted in single PCR products. An additional three markers were removed due to inconsistent amplification success across all samples. Amplification success of primer pairs is presented in Appendix 3, and summary statistics of each locus are presented in Table 2. To check the identity of each sequenced locus, the consensus sequence of the Sanger reads and their primers were mapped back to their respective pseudoreference and the original Citrus gene. For loci with more internally designed primers (i.e., almost entirely intronic), the primers were mapped to both references. In all cases, either the CDS region of the Sanger sequence read itself or the primers aligned to their respective references.
Table 1.
Primer sequences for the nine putative single-copy nuclear markers developed to amplify across Paullinieae (Sapindaceae).

Table 2.
Summary statistics of the nine putative single-copy nuclear markers developed to amplify across Paullinieae (Sapindaceae).a

Phylogenetic analysis—The concatenated alignment of nine markers with a total of 4892 aligned characters was used to generate phylogenetic hypotheses under maximum likelihood. Gaps and the ends of shorter sequences were treated as missing data. Maximum likelihood trees were generated with the general time-reversible (GTR) sequence evolution model in RAxML-HPC on XSEDE 8.2.8 (Stamatakis, 2014) using the CIPRES Scientific Gateway (Miller et al., 2010) (Fig. 2). Support was evaluated with 100 bootstrap replicates. Additionally, gene trees were generated using the GTRGAMMA model in RAxML with 1000 bootstraps on XSEDE 8.2.8 (Stamatakis, 2014). These gene trees were used as input into statistical binning (Mirarab et al., 2014), after which final gene trees were run under a partitioned RAxML run and were used as input in Astral-II ( Appendix S1 (apps.1700051_s1.pdf)). The two liana genera (Paullinia and Serjania) each form monophyletic groups with moderate to high bootstrap support. Multiple individuals of P. turbacensis and P. bracteosa were included in the tree and formed monophyletic groups. The long branch of the Paullinia–Serjania group is explained by the relatively distantly related outgroup A. psilospermus. The Astral-II optimal tree differed from the RAxML tree only in the order of the three P. turbacensis specimens (Fig. 2), providing evidence that these markers are of appropriate length to be informative. Given the sequence variation, moderate to high bootstrap support across nodes, and recovery of monophyly of major groups, we expect these markers to be highly informative with more inclusive sampling.

CONCLUSIONS
This bioinformatic pipeline utilizes publicly available genomic and transcriptomic resources to design primers in coding sequences flanking targeted introns of single-copy nuclear loci without generating sequence data de novo. An annotated genome provides the information to determine approximate intron size and location, and two more closely related transcriptomes provide the best estimate of gene sequence to optimize primer design. By using this combination of data and through primer validation for exclusively single-band PCR products, we increase the chances of targeting and successfully amplifying orthologous single-copy nuclear introns in the lineage of interest. Although many steps were taken to obtain orthologous loci (i.e., only included genes that BLAST exclusively to self, only proceeded with markers that yielded single PCR products, only proceeded with markers that sequenced a single PCR product, and finally mapped Sanger sequences and primers back to their respective pseudoreference and original Citrus gene), orthology could also be further assessed by constructing and analyzing gene trees with greater taxon sampling.
It is important to note that the number of potential target loci can easily be increased by customizing and relaxing parameters or filters throughout the pipeline (e.g., filters for single isoform genes, average gene coverage, desired intron length). By using the closest sequenced genome available, we had the best estimate of intron size and location. This method also worked in Zingiberales (Sass et al., 2016) for designing markers from individuals distant from the genome by approximately 100 mya. Together with increasing transcriptome availability from publicly available sequencing projects like the 1KP project, this pipeline might be increasingly available for many different plant groups. However, if reasonably close genomic and/or transcriptomic resources are unavailable, MarkerMiner (Chamala et al., 2015) is an easy-to-use alternative tool to develop primers spanning introns without the need for a closely related genome. Key differences between MarkerMiner and the bioinformatic pipeline presented here include our use of transcriptome reads rather than a full transcriptome assembly, explicitly targeting introns of a desired size, and the intentional use of the closest available genome.
Several authors present methods to target low-copy nuclear markers for phylogenetic utility (Duarte et al., 2010; Curto et al., 2012; Tonnabel et al., 2014; Stockenhuber et al., 2015; Sass et al., 2016). Using published genomes from across angiosperms, Duarte et al. (2010) recovered 959 genes that were determined to be single copy across Arabidopsis thaliana (L.) Heynh., Populus trichocarpa Torr. & A. Gray, Oryza sativa L., and Vitis vinifera L. (APOV). Interestingly, of these 959 APOV genes, only 201 were found in the C. sinensis genome and, of those, only 24 were determined to be single copy under our criteria. When specifically looking for those 24 remaining single-copy genes following our developed pipeline, we determined that 23 of the 24 APOV genes were removed through various steps specific to our filtering pipeline (i.e., intron size-specific filters, 20× coverage filter, Repeat-Masker filter). Considering copy number in A. thaliana as a reference, Curto et al. (2012) successfully developed nuclear markers in Lamiaceae; however, given the low number of singlecopy APOVs found in C. sinensis due to lineage-specific gene loss and duplication, the APOV markers were not appropriate for our purposes. By using phylogenetically closer genomic and transcriptomic data, we were able to test all markers for copy number prior to including them in phylogenetic analysis. Stockenhuber et al. (2015) and Tonnabel et al. (2014) efficiently detected lowcopy nuclear markers for Brassicaceae and Proteaceae; however, these authors generated transcriptome sequences of members of the lineage of interest. Sass et al. (2016) detected low-copy nuclear markers using publicly available data but targeted exons. The pipeline presented here is cost efficient in that it does not generate sequence data de novo. Rather, it utilizes publicly available genomic and transcriptomic resources spanning the breadth of the plant order Sapindales to design intron markers at conserved coding sequence boundaries. Using the presented pipeline, amplification of nine novel primer pairs was successful in generating phylogenetically informative markers from nine Sapindaceae species, including amplification in the designated outgroup. Sequence variation within these markers ranges from 53.7–94.3% pairwise identity, making them promising for generating a robust data matrix to resolve species-level phylogenetic relationships within Paullinieae, especially when combined with other highly variable markers (e.g., ITS). This flexible marker development pipeline could be applied to any group with appropriate genomic resources. Identified regions of interests can be used in a variety of ways—amplified by PCR and sequenced using Sanger sequencing or as baits for a Hyb-Seq next-generation sequencing approach.
ACKNOWLEDGMENTS
This research was funded by the National Science Foundation (DEB 1208666 to C.D.S.) and by a National Science Foundation Graduate Student Research Grant to J.G.C.
LITERATURE CITED
Appendices
Appendix 1.
Description of the bioinformatic pipeline used to isolate single-copy nuclear introns of a desired size by use of a genome and two sets of transcriptome reads, followed by the primer design and primer testing protocols. All scripts are included in the corresponding directory on https://github.com/joycechery/Sapindaceae or the link is provided in the appendix text. In addition, command syntax for external software, a bash command file, and a description of a process are given.

Continued