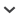Targeted sequence capture, or target enrichment, has emerged as an efficient, cost-effective method for generating phylogenomic data sets for nonmodel organisms (Cronn et al., 2012). The procedure works by reducing genomic DNA complexity through the use of short (80 to 120 nucleotide) bait sequences that hybridize with template sequences. By selectively retaining only genomic fragments bound to baits, high-throughput sequencing libraries are enriched for target sequences. Many samples may be multiplexed and sequenced together, and target enrichment has the potential to generate DNA sequence for hundreds of loci and dozens of samples simultaneously. The methods for generating enriched libraries have been extensively described elsewhere (e.g., Gnirke et al., 2009; Mamanova et al., 2010).
Several recent papers have demonstrated the efficacy of target enrichment to resolve relationships in a variety of organisms (Mandel et al., 2014; Mariac et al., 2014; Bragg et al., 2015). In one strategy, ultra-conserved elements can be used to anchor baits in slow-evolving portions of the genome, and analysis is focused on more variable flanking regions (Faircloth et al., 2012; Lemmon et al., 2012). Another approach is to focus on exon sequences, because reference sequences across phylogenetic scales can be efficiently generated using transcriptome sequencing (Bi et al., 2012; Hugall et al., 2016). An extension of this approach is Hyb-Seq (Weitemier et al., 2014), which combines exon capture with genome skimming of a “splash zone”— intronic and intergenic regions that flank target exons, potentially of use for shallower phylogenetic applications.
In previously published studies using Hyb-Seq data, three main bioinformatics issues have arisen: (1) how to efficiently sort high-throughput sequencing reads into separate loci (e.g., Stull et al., 2013), (2) how to assemble sequences at each locus that can be aligned for phylogenetic inference (e.g., Stephens et al., 2015), and (3) how to extend sequence recovery beyond the coding sequence into the more variable intron regions (e.g., Folk et al., 2015). Data need to be handled in an efficient, streamlined manner because many Hyb-Seq projects involve dozens or hundreds of samples.
We developed HybPiper to efficiently turn sequencing reads generated by the Hyb-Seq method into organized gene files ready for phylogenetic analysis. HybPiper is a suite of Python scripts that wrap and organize bioinformatics tools for target sequence extraction from high-throughput sequencing reads. The primary output of the pipeline is a nucleotide and translated amino acid sequence for every gene that can be assembled from the sequencing reads. HybPiper also includes several postprocessing scripts for retrieving sequences from multiple samples run through the pipeline, visualization of summary statistics such as recovery efficiency and coverage depth, and extraction of flanking intron sequences. We designed the pipeline to be easy-to-use in a modular design that allows the user to rerun portions of the pipeline to adapt parameter settings (i.e., E-value thresholds, assembly coverage cutoffs, or percent identity filters) for individual samples as needed. Although other bioinformatics pipelines are available to process target enrichment data, such as PHYLUCE (Faircloth, 2015) and alignreads.py (Straub et al., 2011), HybPiper is designed specifically for the Hyb-Seq approach: targeting exons and flanking intron regions.
METHODS AND RESULTS
Input data—Here, we demonstrate the utility of HybPiper using 22 species of Artocarpus J. R. Forst. & G. Forst. (Moraceae) and six outgroups. Sequencing libraries were hybridized to a bait set comprising 458 target nuclear coding regions. We describe the development of bait sequences from a draft genome sequence in a companion paper (Gardner et al., 2016). Briefly, 333 loci intended for phylogenetic analysis were selected by identifying long exons homologous between the Artocarpus draft genome and the published genome of Morus notabilis C. K. Schneid. (Moraceae) (He et al., 2013). For the “phylogenetic genes,” the bait set included 120-bp baits designed from both Artocarpus and Morus sequences. A set of 125 additional genes were targeted for their functional significance: 98 MADS-box genes and 27 genes that have been implicated in floral volatiles. For the genes of functional significance, baits were designed from the A. camansi Blanco draft genome alone (Gardner et al., 2016). A set of 20,000 baits (biotinylated RNA oligonucleotides, the smallest MYbaits kit) with 3× tiling was manufactured by MYcroarray (Ann Arbor, Michigan, USA).
Sequencing libraries for 22 Artocarpus species and two of the outgroups (Table 1) were prepared with the Illumina TruSeq Nano HT DNA Library Preparation Kit (Illumina, San Diego, California, USA) following the manufacturer's protocol, with a target mean insert size of 550 bp. Libraries were hybridized to the bait set in four pools of six libraries each at 65°C for approximately 18 h, following the manufacturer's protocol. The enriched libraries were reamplified with 14 PCR cycles. The four pools of enriched libraries were sequenced together in a single flow cell of Illumina MiSeq (600 cycle, version 3 chemistry). This run produced 9,503,831 pairs of 300-bp reads. Four additional outgroup libraries generated in a separate hybridization were sequenced as part of a separate run and generated an additional 3,716,390 pairs of reads. Demultiplexed and adapter-trimmed reads (cleaned automatically by Illumina BaseSpace) were quality trimmed using Trimmomatic (Bolger et al., 2014), with a quality cutoff of 20 in a 4-bp sliding window, discarding any reads trimmed to under 30 bp. Only pairs with both mates surviving were used for HybPiper. An average of 391,505 reads per sample survived the trimming process across all 28 samples. The reads have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (Bio-Project PRJNA301299).
The inputs of HybPiper are the read file (or files, for paired-end data) and a curated “target file.” HybPiper is built to operate at the locus level; if target sequences were designed from multiple exons within the same gene, these may be concatenated (with no gaps or intervening sequence) to generate a single coding sequence for each gene. This allows HybPiper to detect intron sequences during coding sequence extraction. In the case of the Artocarpus/Morus bait set, two orthologous sequences are retrieved for most loci. The presence of multiple sequences for each locus is specified in the sequence IDs within the target file; for example, “Artocarpus-g001” and “Morus-g001” indicate to HybPiper that both sequences represent locus g001. Phase 1 of HybPiper, in part, determines which sequence is the more appropriate reference sequence for each gene and sample separately. This flexibility allows the use of the same target file for samples that span a wide range of phylogenetic distances. Additional orthologous sequences for each gene may be added to the target file as desired by the user, which may increase the efficiency of sorting reads and generate new orthologous loci for phylogenetics.
Phase 1: Sorting sequencing reads by target gene—Target enrichment is typically conducted on multiple samples that have been pooled during bait hybridization and sequencing. HybPiper maps reads against the target genes for each sample separately. This is a different procedure than several other target enrichment analysis pipelines (Straub et al., 2011; Bi et al., 2012; Faircloth, 2015), which typically begin with de novo assembly for each sample, and then attempt to match contigs to target loci. In HybPiper, reads are first sorted based on whether they map to a target locus. We explored two methods for aligning reads to the targets: (1) BLASTX (Camacho et al., 2009), which uses peptide sequences as a reference, and (2) BWA (Li and Durbin, 2009), which uses nucleotide sequences. In principle, the BLASTX approach should be more forgiving to substitutions between the target sequence and sample reads, because alignments are conducted at the peptide level and may detect similarity between more distant sequences than BWA. The BWA approach may result in fewer overall reads mapping to a distantly related target sequence, but is several times faster than the BLASTX method.
HybPiper sorts reads into separate directories for each gene using Biopython (Cock et al., 2009) to efficiently parse the FASTA format. In our tests of the BLASTX method, we set an E-value threshold of 1 × 10−5 to accept alignments, but the user can change this. For the BWA method, all alignable reads are sorted into each gene directory using a Python wrapper around SAMtools (Li et al., 2009). We calculate the enrichment efficiency as the percentage of trimmed, filtered reads that were sorted into a gene directory.
For the Artocarpus reads, an average of 71.9% of reads were on target (range 64.4–79.9%), based on the BLASTX method. Enrichment efficiency was lower for some of the outgroup samples, which ranged from just 5.0% for Antiaropsis K. Schum. to 71.6% for Ficus L. To address whether the presence of duplicate reads affects our estimate of enrichment efficiency, we removed paired duplicate reads using SuperDeduper ( http://dstreett.github.io/Super-Deduper/). Most samples had between 6% and 18% duplicate read pairs, and a similar percentage of the duplicate read pairs mapped to the target loci ( Appendix S1 (apps.1600016_s1_s2_s3_s5_s6.xlsx)). One outlier was Ficus, which had 34% duplicate reads, 42% of which mapped to targets. After adjusting for duplicate reads, our estimates of enrichment efficiency were reduced by about 4% on average (Table 1). Removing duplicate reads did not affect the extraction of exon sequences in HybPiper for this data set.
The phylogenetic distance to Artocarpus did not seem related to percent enrichment. However, the two outgroup samples that were pooled in a hybridization with Artocarpus in the first sequencing run had much lower enrichment efficiency than ingroup samples (Table 1). This suggests that multiplexing at the hybridization stage should be nonrandom, and only libraries of taxa that are relatively equidistant from the taxa used to design the bait sequences should be pooled. This strategy has been previously recommended in other studies (McGee et al., 2016).
Phase 2: Sequence assembly—Some previous methods for target enrichment assembly have used mapping-based approaches to reassemble target loci (Straub et al., 2011; Hugall et al., 2016), which may be inefficient when there is high sequence divergence between the sample reads and the target reference. HybPiper instead conducts a de novo assembly for each gene separately; reads are assembled into contiguous sequences (contigs) using SPAdes (Bankevich et al., 2012). Multiple contigs may be assembled per gene, due to incomplete sequencing of intron sequences, paralogous gene sequences, or alleles. Additional contigs may be assembled from weakly aligning reads with low identity to the target sequences. These contigs are sorted by sequencing depth and are aligned to the reference protein sequence in the next phase. HybPiper decreases the amount of time needed for assembly and alignment stages by using GNU Parallel, a tool for executing commands simultaneously, using the multiple threads available on modern processors (Tange, 2011).
Phase 3: Alignment of exons—In 333 of 458 genes in our test data set, baits were designed from homologous sequences in the A. camansi draft genome and the previously published M. notabilis genome. For each gene, HybPiper decides whether the Artocarpus or Morus target sequence should serve as the reference by tallying all alignment scores from reads aligned to the gene during Phase 1. In the BWA version of the alignment, the “mapping score” is tallied for all reads mapping to the target gene; for the BLASTX method, the bit score is used.
Target enrichment is generally carried out using genomic DNA; however, bait sequences are often designed from only the coding regions of a target, such as assembled transcripts. To extract the coding sequence portion of the assembled contigs that likely contain partial intron sequence, HybPiper uses Exonerate (Slater and Birney, 2005). For each target, assembled contigs are aligned to target peptide sequences using the “protein2genome” alignment model. If the BWA method was used for alignment, the peptide sequence is generated by direct translation using Biopython. Sample sequences homologous to the reference coding sequence are extracted in FASTA format with a customized header specifying alignment start and end locations and percent identity between the sample and reference, using the “roll your own” feature in Exonerate.
Table 1.
Sample information, sequencing run and hybridization pool, summary of sequencing, and target enrichment results for the Artocarpus/Morus bait set.

Within HybPiper, Exonerate (Slater and Birney, 2005) is used to extract likely coding sequences (introns removed) aligned to the reference protein sequence. These alignments must be nonoverlapping and exceed a percent identity threshold (default: 60%) between the contig and the protein sequence. Alignments are sorted by position (relative to the reference sequence), and the longest contig that does not overlap with other contigs is retained. However, if the overlap between the ends of two contig alignments is less than 20 bp, both contigs are retained. This is to reduce errors in alignment at the ends of exons. Any contigs with slightly overlapping ranges are concatenated into a “supercontig” and a second Exonerate analysis is conducted to detect the true intron-exon junctions. At this stage, the coding sequence that aligns to the reference amino acid sequence and statistics about the contigs retained are saved into the gene sequence directory.
Identification of paralogous sequences, alleles, or contaminants—In many target enrichment analysis pipelines, correct orthology of enriched sequences is inferred using BLAST searches to the target proteome (e.g., Bi et al., 2012; Bragg et al., 2015), but this method will be inefficient when genomic resources in the target taxa are incomplete. In HybPiper we provide a streamlined method for identification of potential paralogs that can be further analyzed using gene phylogenies. Typically, if HybPiper identifies a single contig that subsumes the range of other contigs, it is retained and the smaller contigs are discarded. However, sequences assembled using SPAdes occasionally result in multiple, long contigs, each representing the entire target sequence. During the extraction of exon sequences, the HybPiper script exonerate_hits.py identifies contigs that span more than 85% of the length of the reference sequence. HybPiper will generate a warning that indicates multiple long-length matches to the reference sequence have been found. HybPiper chooses among multiple full-length contigs by first using a sequencing coverage depth cutoff—if one contig has a coverage depth 10 times (by default) greater than the next best full-length contig, it is chosen. If the sequencing depth is similar among all full-length contigs, the percent identity with the reference sequence is used as the criterion. Genes for which multiple long-length sequences exist should be examined further to detect whether they represent paralogous genes, alleles, or contaminants. We discuss identification of paralogs in more detail below (see “Separating paralogous gene copies in Artocarpus”).
Fig. 1.
Heat map showing recovery efficiency for 458 genes enriched in Artocarpus and other Moraceae and recovered by HybPiper using the BWA method. Each column is a gene, and each row is one sample. The shade of gray in the cell is determined by the length of sequence recovered by the pipeline, divided by the length of the reference gene (maximum of 1.0). Three types of genes were enriched: phylogenes (left), MADS-box genes (center), and volatiles (right) for 22 Artocarpus samples (top) and six outgroup species (bottom). Full data for this chart can be found in Appendices S2 and S3 (apps.1600016_s1_s2_s3_s5_s6.xlsx).

Extraction of flanking intron sequences—Following the identification of exon sequences in the assembled contigs, HybPiper attempts to identify intron regions flanking the exons using the script “intronerate.py.” This is done by rerunning Exonerate on the supercontigs used in Phase 3 and retaining the entire gene sequence, rather than just the exon sequence. Even if the entire intron was not recovered, Exonerate can detect the presence of splice junctions based on the alignment of the supercontig to the reference protein sequence. HybPiper generates an annotation of the supercontig in genomic feature format (GFF). The annotations are sorted and filtered in the same manner as the exon sequences during Phase 3 (alignment of exons) of the main HybPiper script, to remove overlapping annotations. Following the intron annotation, HybPiper also produces two additional sequence files at each locus: (1) the supercontig and (2) only the intron sequences (exons removed from supercontig).
Postprocessing: Visualization of recovery efficiency—After running HybPiper on multiple samples, we have provided a series of helper scripts to collect and visualize summary statistics across samples. Running these scripts from a directory containing all of the output of each sample takes advantage of the standardized directory structure generated by HybPiper. The script get_gene_lengths.py will summarize the length of the sequences recovered and will return a warning if a sequence is more than 50% longer than the corresponding target sequence. This file can be used as the input for an R script included with HybPiper (gene_recovery_heatmap.R) to visualize the recovery efficiency using a heat map (Fig. 1).
In the heat map, each row represents a sample, and each column represents a target. Each cell is shaded based on the length of the sequence recovered by HybPiper for that gene, as a percentage of the length of the reference sequence. The heat map can be used as a first glance at the efficiency of target recovery and help identify difficult-to-recover loci (columns with lighter shading) and low-enrichment samples (rows with lighter shading).
For the Artocarpus data set, we were able to recover the vast majority of loci for in-group samples (Table 2). Using 10 processors on a Linux computer, HybPiper completed in about 9 h using the BWA method and 24 h using BLASTX for all 28 samples. For all samples, including outgroups, HybPiper was able to recover the 333 “phylogenetic genes” with more efficiency than the MADS-box or “volatile” genes. For instance, HybPiper recovered 93% (311 of 333) of the phylogenetic loci for Ficus, but just 30% (37/125) of the MADS-box and volatile loci using the BWA method (Table 2). The most likely explanation for this is that the phylogenetic loci had bait sequences derived from two different sources (Artocarpus baits and Morus baits). The remaining 125 loci had baits designed only from Artocarpus draft genome sequence. Substitutions between the Artocarpus sequences and our outgroup samples may have reduced the hybridization efficiency for the MADS-box and volatile genes. The dissimilarity between target sequence and sample sequences was compounded by using the BWA method, which allows for less dissimilarity than the BLASTX method for aligning reads (Table 2). Additional information about the recovery of loci using the BLASTX and BWA methods can be found in the supplemental material ( Appendices S2 and S3 (apps.1600016_s1_s2_s3_s5_s6.xlsx)).
Table 2.
Recovery efficiency of HybPiper for 22 Artocarpus and six other Moraceae, using two methods for assigning reads to loci—BLASTX (mapping to protein sequences) and BWA (mapping to nucleotide sequences).

Alternatively, the increased hybridization efficiency for the phylogenetic genes may be the result of twice as many bait sequences for each target. The pooling strategy, during hybridization and sequencing, may have also had an effect. In our first sequencing run, the two outgroup species had about one third of the average number of reads, and about one tenth the average number of reads on target, compared to the Artocarpus species. Therefore, pooling outgroup samples together in separate hybridizations may be advisable in future Hyb-Seq analyses.
Recovery of intron sequences in Artocarpus—The utility of phylogenomic approaches is maximized when the resolution at individual loci is sufficient for the phylogenetic depth of the analysis (Salichos and Rokas, 2013). This is especially true for “species tree” methods that require gene tree reconstructions as input (Mirarab et al., 2014). For adaptive radiations and species complexes, ultraconserved elements may not be variable enough to resolve internodes with only a few parsimony informative characters per locus (Smith et al., 2013; e.g., Giarla and Esselstyn, 2015; Manthey et al., 2016). Newer sequencing technologies, such as the 2 × 300 (paired-end) MiSeq chemistry from Illumina, produce reads that are longer than the typical bait length, resulting in sequence fragments containing pieces of exon (i.e., on-target) that may also extend hundreds of base pairs into intron or intergenic regions. This is an attractive solution because the same bait sets designed for deeper phylogenetic questions, where exon variability may be sufficient, could also be used at shallower scales.
We explored the capture efficiency of intron regions in the Artocarpus bait set by aligning reads of Artocarpus samples to the reference genome scaffolds using BWA. Two patterns emerged: for short introns (<500 bp), little difference was detected in the depth of coverage between exons and introns (Fig. 2). For longer introns, depth steadily decreased but was typically still above 10× up to 500 bp away from the end of the exon (Fig. 2), even with duplicate reads removed ( Appendix S4 (apps.1600016_s4.pdf)). This suggests that long-read technologies such as MiSeq 2 × 300 paired-end sequencing are well-suited for recovering intronic regions using Hyb-Seq.
Recent studies have explored the feasibility of using introns extracted from Hyb-Seq (Folk et al., 2015; Brandley et al., 2015), but have not presented an automated method for extracting intron sequence from capture data. HybPiper can generate “supercontigs” containing all assembled contigs (exon and intron sequence) for a gene. This sequence is annotated in genome feature format (GFF), which can be used to extract intronic and/or intergenic regions into separate files for alignment and analysis. We observe the greatest reliability of extracting intronic regions by generating multiple sequence alignments of the supercontigs from multiple samples. The exon regions serve as an anchor for the alignment, and extraneous sequence that appears in only one or a few sequences can be trimmed by downstream analysis, such as trimAl (Capella-Gutierrez et al., 2009).
For the 333 loci developed as phylogenetic markers, extracting intron data vs. exon data alone using HybPiper increased the average length of the loci from 1135 bp (range 201–3171 bp) to 1784 bp (range 528–4267 bp) ( Appendix S5 (apps.1600016_s1_s2_s3_s5_s6.xlsx)). When all samples were aligned using MAFFT and trimmed using trimAl, the total alignment length increased to 594,149 bp and added 138,982 characters to the concatenated alignment. The number of parsimony informative characters within Artocarpus increased from 35,935 using exons only to 138,932 using supercontigs. Intron sequence recovery efficiency was variable across the loci; for 54 loci the full alignment length was within 100 bp of the exon-only alignment. In contrast, 172 loci had a final alignment length 500 bp or longer than that of exons alone.
Separating paralogous gene copies in Artocarpus—The genus Artocarpus has undergone at least one whole genome duplication since its divergence from the rest of the Moraceae (Gardner et al., 2016). As a result, many genes that appear single copy in the Morus reference genome are multicopy in Artocarpus. HybPiper identified paralogous gene copies in Artocarpus for 123 of our 333 phylogenetic loci ( Appendix S6 (apps.1600016_s1_s2_s3_s5_s6.xlsx)). For these genes, multiple full-length contigs were assembled with similar sequencing coverage. To investigate the paralagous copies further, we extracted the paralogous exon sequence from each contig using two scripts included in HybPiper: paralog_investigator.py identifies whether multiple contigs that are at least 85% of the target reference length are present and flags these as possible paralogs. It then extracts exon sequences from putative paralogs using exonerate_hits.py. A second script, paralog_retriever.py, collects the inferred paralogous sequences across many samples for one gene. If no paralogs are identified for a sample, the coding sequence extracted during Phase 3 of the main script is included.
We generated gene family phylogenies for several genes where multiple copies were identified in Artocarpus. Nucleotide sequences were aligned using MAFFT (Katoh and Standley, 2013) (--localpair --maxiterate 1000) and phylogenies were reconstructed using RAxML using a GTRGAMMA substitution model and 200 “fast-bootstrap” pseudoreplicates. In each case, two separate clades of Artocarpus samples are observed ( Appendix S7 (apps.1600016_s7.pdf)), indicating that the multiple full-length contigs likely result from the paleopolyploidy event, and that they can be distinguished by HybPiper. For further phylogenetic analysis, the paralog with the highest percent identity to the Artocarpus camansi reference genome sequence was selected for each sample, because this paralog represents the closest homology to the sequence from which the baits were designed.
Phylogeny reconstruction—After running HybPiper on multiple samples, multisequence FASTA files can be generated for each gene using a script included with the pipeline (retrieve_sequences.py). After aligning each gene separately with MAFFT, we reconstructed phylogenies with two nucleotide supermatrix data sets, (1) the full matrix and (2) the exons alone, in RAxML (Stamatakis, 2014) using a separate GTRGAMMA partition for each locus, along with 200 “fast-bootstrap” pseudoreplicates.
Fig. 2.
Depth-of-coverage plots for four exemplar loci based on reads aligned to the Artocarpus camansi draft genome. Each gray line represents a rolling average depth across 50 bp for one of 22 Artocarpus species. The dark line represents the average depth of coverage. Red bars indicate the location of exon boundaries predicted in the Artocarpus camansi draft genome.

The phylogenies are largely similar in topology and level of support ( Appendix S8) (apps.1600016_s8.pdf), particularly for most subgenera circumscribed by Zerega et al. (2010). Rearrangements or changes in bootstrap support are occasional and minor. Only one rearrangement was characterized by high support in both positions (A. limpato Miq.). Although the phylogenies are for the most part well resolved, they should be treated with caution due to the low taxon sampling (only 22 out of ca. 70 species). We recognize here that using a supermatrix approach alone is insufficient to fully understand the influence of conflicting gene-tree signal due to processes such as incomplete lineage sorting. We present, however, this phylogeny simply as an example of one possible analysis that can be performed using the output of HybPiper. The output files generated by HybPiper are suitable for use with whichever phylogeny reconstruction method is favored by the user.
CONCLUSIONS
HybPiper can be used to efficiently assemble gene regions from enriched sequencing libraries designed using the Hyb-Seq method, extract exon and intron sequences, and assemble sequence data that are ready to use in phylogenetic analysis. The pipeline is flexible and modular, and can be adapted to analyses at deep phylogenetic depths (by using the BLASTX method) or within genera (by incorporating intron sequence). The pipeline has been tested on Linux and Mac OS X, and is freely available under a GPLv3 license at: https://github.com/mossmatters/HybPiper.
ACKNOWLEDGMENTS
We would like to thank A. DeVault at MycroArray for assistance optimizing the target enrichment protocol, and the Field Museum for use of its DNA sequencers. The authors thank B. Faircloth and two anonymous reviewers for helpful comments on an earlier version of the manuscript. This research was funded by National Science Foundation grants to A.J.S. (DEB-1239980), B.G. (DEB-1240045 and DEB-1146295), N.J.W. (DEB-1239992), and N.J.C.Z. (DEB-0919119), and by a grant from the Northwestern University Institute for Sustainability and Energy (N.J.C.Z.). Data generated for this study can be found at www.artocarpusresearch.org, www.datadryad.org( http://dx.doi.org/10.5061/dryad.3293r), and the NCBI Sequence Read Archive (SRA; BioProject PRJNA301299).