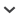The importance of incorporating low-copy nuclear genes in phylogenetic reconstruction is well-recognized, but has largely been constrained by technical limitations (Zimmer and Wen, 2013). These data are essential for reconstructing the evolutionary history of plants, including understanding the causes of observed incongruities among gene trees that arise from incomplete lineage sorting and introgressive hybridization. The combination of solution hybridization for target enrichment of specific genomic regions and the high sequencing throughput of current platforms (e.g., Illumina) provides the opportunity to sequence hundreds or thousands of low-copy nuclear loci appropriate for phylogenetic analyses in an efficient and cost-effective manner (Cronn et al., 2012; Lemmon and Lemmon, 2013). Most efforts to date for targeted sequencing of plant genomes for phylogenetics have been directed at the plastome (e.g., Parks et al., 2012; Stull et al., 2013). Recently, conserved orthologous sequences in Asteraceae (Chapman et al., 2007) were obtained via target enrichment for phylogenomics (Mandel et al., 2014).
Methods have been developed to target highly or ultra-conserved elements (UCEs) in animal genomes (Faircloth et al., 2012; Lemmon and Lemmon, 2013; McCormack et al., 2013). However, UCEs in plants are nonsyntenic, and are hypothesized to have originated via horizontal transfer from organelles or de novo evolution (Reneker et al., 2012). Whatever their origin, their potential for nonorthology among species makes them unsuitable as phylogenetic markers in plants. The frequency of polyploidy throughout angiosperm evolution (Jiao et al., 2011) also impedes obtaining a large set of conserved orthologous single-copy loci transferable across plant lineages, which in combination with the lack of orthologous UCEs, means that design of targeted sequencing strategies for plant nuclear genomes will necessarily be lineage-specific.
Here we present Hyb-Seq, a protocol that combines target enrichment of low-copy nuclear genes and genome skimming (Straub et al., 2012), the use of low-coverage shotgun sequencing to assemble high-copy genomic targets. Our protocol improves upon the methods of Mandel et al. (2014) by (1) utilizing the genome and transcriptome of a single species for probe design, which makes our approach more generally applicable to any plant lineage; (2) obtaining additional data from the procedure through combination with genome skimming; and (3) developing a data analysis pipeline that maximizes the data usable for phylogenomic analyses. Furthermore, we assemble sequences from the flanking regions (the “splash zone”) of targeted exons, yielding noncoding sequence from introns or sequence 5′ or 3′ to genes, which are potentially useful for resolving relationships at low taxonomic levels. We demonstrate the feasibility and utility of Hyb-Seq in a recent, rapid evolutionary radiation: Asclepias L. (Apocynaceae). Target enrichment probes were designed using the A. syriaca L. draft genome and transcriptome sequences in concert to identify nuclear loci of sufficient length (>960 bp) for robust gene tree reconstruction with a high probability of being single copy (>10% divergence from all other loci in the target genome). We also demonstrate the utility of the Asclepias data for phylogenomic analysis and explore the utility of the probes for Hyb-Seq in another genus of the same subtribe, Calotropis R. Br. (Asclepiadineae), and a more distantly related genus, Matelea Aubl. (Gonolobineae). The Hyb-Seq approach presented here efficiently obtains genome-scale data appropriate for phylogenomic analyses in plants, and highlights the utility of extending genomic tools developed from a single individual for use at deeper phylogenetic levels.
METHODS AND RESULTS
Targeted enrichment probe design—An approach was developed for Hyb-Seq probe design (Table 1) in Asclepias utilizing a draft assembly of the A. syriaca nuclear genome (Weitemier et al., unpublished data), which was assembled using Illumina paired-end data from libraries with insert sizes of 200 and 450 bp and a k-mer size of 79 in ABySS v. 1.3.2 (Simpson et al., 2009) with reads of plastid or mitochondrial origin removed prior to assembly. A transcriptome of A. syriaca leaf and bud tissue (Straub et al., unpublished data) was assembled de novo using Trinity RNA-Seq v. r20131110 (Grabherr et al., 2011) and refined using transcripts_to_best_scoring_ORFs. pl (included with Trinity). Probe design was based on data from the draft genome, which was combined with transcriptome assembly data to target the exons of hundreds of low-copy loci. Contigs from the draft nuclear genome were matched against those sharing 99% sequence identity from the transcriptome using the program BLAT v. 32 × 1 (Kent, 2002). BLAT accommodates large gaps in matches between target and query sequences, and is suitable for matching the exon-only sequence of transcripts with the intron-containing genomic sequence, allowing the locations of potential intron/exon boundaries to be identified. Additionally, in an effort to prevent loci present in multiple copies within the genome from being targeted, only those transcripts with a single match against the genome were retained. To prevent probes from enriching multiple similar loci, any targets sharing ≥90% sequence similarity were removed using CD-HIT-EST v. 4.5.4 (Li and Godzik, 2006). The remaining transcriptome contigs were filtered to retain only those containing exons ≥120 bp totaling at least 960 bp. The lower cutoff was necessary to provide sufficiently long sequences for probe design (=120 bp), and the upper cutoff was chosen to exclude short loci less likely to include phylogenetically informative sites. Of the loci that passed filtering, all of those matching (70% sequence identity over 30% of its length) a previously characterized putative ortholog from Apocynaceae (expressed sequence tags [ESTs] from Catharanthus roseus (L.) G. Don; Murata et al., 2006), the asterids (COSII; Wu et al., 2006), or four nonasterid angiosperms (Duarte et al., 2010) were retained (1335 exons in 350 loci). Additional loci that passed filtering were added to the set of targeted loci until the total length of the target probes approached the minimum required for oligonucleotide synthesis (2050 exons in 418 loci). The final probe set also contained probes intended to generate data for other projects (157 defense-related and floral development genes and 4000 single nucleotide polymorphisms [SNPs]), which were only included here where necessary for calculations of hybridization efficiency and assembly length. Note that care should be taken during the probe design process to avoid targeting organellar sequences together with nuclear sequences, because enrichment of organellar targets will be proportional to their presence in the genomic DNA extractions used to prepare sequencing libraries and may greatly exceed nuclear targets (see Appendix S1 (apps.1400042_s1.pdf)).
Table 1.
Hyb-Seq target enrichment probe design and bioinformatics pipeline. A script combining and detailing the steps of the probe design process, Building_exon_probes.sh, is provided in the supplementary materials ( Appendix S1 (apps.1400042_s1.pdf)).

Illumina library preparation and Hyb-Seq—DNA was extracted from 10 species of Asclepias, Calotropis procera (Aiton) W. T. Aiton, and Matelea cynanchoides (Engelm. & A. Gray) Woodson (Appendix 1) using either a modified cetyltrimethylammonium bromide (CTAB) protocol (Doyle and Doyle, 1987), DNeasy (QIAGEN, Valencia, California, USA), FastDNA (MP Bio, Santa Ana, California, USA), or Wizard kits (Promega Corporation, Madison, Wisconsin, USA). Most indexed Illumina libraries were prepared as described by Straub et al. (2012). Two exceptions were A. cryptoceras S. Watson (prepared with a NEXTflex DNA barcode; Bioo Scientific, Austin, Texas, USA) and M. cynanchoides (TruSeq library preparation kit; Illumina, San Diego, California, USA). Libraries were then pooled in 11- or 12-plexes with approximately equimolar ratios (some samples included in the pools were not included in the current study). Solution hybridization with MYbaits biotinylated RNA baits (MYcroarray, Ann Arbor, Michigan, USA) and enrichment followed Tennessen et al. (2013) with approximately 350–480 ng of input DNA per pool and 12 rather than 15 cycles of PCR enrichment to decrease the production of PCR duplicates. These target-enriched libraries were then sequenced on an Illumina MiSeq at either Oregon Health Science University (version 2 chemistry) to obtain 2 × 251-bp reads or Oregon State University (version 3 chemistry) to obtain 2 × 76-bp reads. Raw Illumina data were submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRP043058).
Data analysis pipelines—Raw data were filtered for adapter sequences either by the sequencing centers, using Trimmomatic v. 0.20 or 0.30 (Bolger et al., 2014), or using custom scripts. Internal sequence barcodes were deconvoluted using bc_sort_pe.pl (Knaus, 2012). Reads were quality filtered using Trimmomatic to remove bases at read ends with qualities lower than Q20, to trim the rest of the read when average quality in a 5-bp window was <Q20, and to remove reads shorter than 36 bp following trimming. For A. cryptoceras, only read ends were trimmed to Q20. Duplicate reads were removed using the FASTX-Toolkit (Gordon, 2010). For target assembly, a reference-guided approach utilizing a pseudo-reference consisting of targeted exons separated by 200 Ns was implemented in Alignreads v. 2.25 (Straub et al., 2011). BLAT was used to identify contigs in the final assembly with sequence similarity to targeted exons. A custom script extracted the longest assembled sequence corresponding to each exon and constructed matrices for multiple sequence alignment, while adding Ns to the matrix if an exon was missing for a particular species. Exons were aligned using default settings in MAFFT v. 6.864b (Katoh and Toh, 2008). Following alignment, exons of the same gene were joined using catfasta2phyml.pl. Splash-zone sequences were not included in this analysis. The same read pools were then used for reference-guided assembly of high-copy sequences, the plastome and nuclear ribosomal DNA (nrDNA) cistrons (18S-5.8S-26S), using Alignreads. The references used for each species were generated through analysis of previously collected genome skim data of the same libraries used for this study (Straub et al., 2012; Straub et al., unpublished data). References from a different A. cryptoceras individual were used for that species and reads were retrimmed using the Trimmomatic setting described above. The M. biflora (Raf.) Woodson plastome (GenBank: KF539850.1) and the C. procera nrDNA sequence served as references for M. cynanchoides. MAFFT was used for alignment. Appendix S2 (apps.1400042_s2.pdf) provides additional details on bioinformatic analyses.
Analyses of assembled sequences—The total length of assembled sequence, numbers of targeted exons and genes assembled, amount of flanking sequence assembled from the splash zone, percentage of plastome and nrDNA cistron sequence assembled from the off-target reads based on the lengths of the reference sequences, and percent divergence from the A. syriaca exon sequences were calculated for each species. The Hyb-Seq data were also analyzed using the phyluce v. 1.4 pipeline (Faircloth et al., 2012; Faircloth, 2014) used by Mandel et al. (2014), using both the native de novo assembly option and using the contigs produced by the reference-guided assembly in Alignreads as input data (see Appendix S3 (apps.1400042_s3.pdf) for detailed methods). To demonstrate the utility of the data for phylogenomics, analyses of Asclepias and outgroup Calotropis were conducted for nuclear genes individually (excluding seven genes with terminals with all missing data), a concatenation of all nuclear genes, and whole plastomes using RAxML v. 7.3.0 (Stamatakis, 2006) with a GTR + Γ model of nucleotide substitution. One hundred and 1000 rapid bootstrap replicates were conducted for nuclear and plastid analyses, respectively. Prior to analysis, the plastome matrix was edited following Straub et al. (2012). RAxML nuclear gene trees were then used for phylogenomic analyses of all targeted loci with complete taxon sampling (n = 761) using the MP-EST species tree approach with bootstrap evaluation of clade support implemented through the STRAW Webserver (Shaw et al., 2013). Targeted nuclear exon sequences, data matrices, and trees were submitted to Figshare ( http://dx.doi.org/10.6084/m9.figshare.1024614). See Appendices S1 (apps.1400042_s1.pdf) and S2 (apps.1400042_s2.pdf) for detailed discussion of the protocol from probe design to data analysis.
Hyb-Seq results—We identified 768 putatively single-copy genes (3385 exons, ca. 1.6 Mbp) meeting the criteria of sufficient length and divergence from all other genes in the genome. Of these genes, 136 genes were among asterid COSII sequences and 42 were among genes conserved across four angiosperm genomes; only 12 out of 155 possible overlapping genes were shared by both conserved sets. Enrichment, sequencing, and assembly of the targeted putatively single-copy genes was successful in Asclepias and related Apocynaceae with at least partial assembly of an average of 92.6% of exons and 99.7% of genes and a total average assembly length for all genes in the probe set of ca. 2.2 Mbp from 1.7 Mbp of targeted exons (including the defense and floral development genes; Table 2). Lower read numbers (due to unequal library pooling) resulted in reduced target capture and assembly efficiency in A. eriocarpa Benth. and A. involucrata Engelm. ex Torr. (Table 2), while a combination of lower read number and sequence divergence (average 4.5%) between Matelea and the probes is likely responsible for its somewhat lower success (Fig. 1; Table 2). In contrast, target capture in Calotropis (average 3.2% divergence from A. syriaca) was similar to Asclepias (Table 2). Given that the probes should work well up to 10% sequence divergence, this probe set is likely useful for enrichment of the targeted genes across Asclepiadoideae (Fig. 1). Extending the comparison to the more distantly related Catharanthus roseus (Gongora-Castillo et al., 2012), BLAT analysis reveals an average 12% divergence between A. syriaca exons and orthologous transcripts (Fig. 1). This result predicts that a smaller, but not insignificant, amount of sequence data could be obtained from the rest of Apocynaceae. Modification of hybridization conditions could further increase success for more divergent species (Li et al., 2013). In addition to the targeted nuclear loci, reference-guided assembly of the off-target reads yielded complete or nearly complete plastome and nrDNA cistron sequences (Table 2).
Table 2.
Success of Hyb-Seq for targeted sequencing and assembly of nuclear genes combined with genome skimming of high-copy targets in Asclepias and related species of Apocynaceae.

The data analysis pipeline presented here resulted in a data set with few missing genes for each species. In contrast, the phyluce pipeline recovered comparatively few loci for phylogenomic analysis (Table 3). Phyluce was designed for the analysis of UCE data, and its adoption for analysis of single-copy genes where multiple exons have been targeted is inappropriate because exons are often assembled on separate contigs and phyluce views multiple contigs matching a targeted locus as an indication of paralogy (see Appendix S3 (apps.1400042_s3.pdf) for further discussion). The use of reference-guided assembly in the pipeline presented here, rather than the de novo approach of phyluce, also results in a greater amount of data recovery for use in phylogenomic analyses (Table 3).
Phylogenomics—Percentage of variable sites within 768 sequence alignments ranged from 1.8% to 12.5%, with a mean of 5.9% ( Appendix S4 (apps.1400042_s4.pdf)). The concatenated data matrix was 1,604,805 bp, with 104,717 variable sites, 10,210 of which were parsimony informative. Phylogenomic analysis of the maximum likelihood gene trees for the 761 putatively single-copy genes containing information for all 12 taxa resulted in a species tree topology in which most nodes received high bootstrap support, and which differed from the concatenation species tree in the placement of A. eriocarpa (Fig. 2, left). This result highlights the importance of utilizing species tree methods and approaches for assessing clade support that take into account discordance among gene trees, because concatenation approaches can result in strongly supported, but misleading inferences of evolutionary relationships (Kubatko and Degnan, 2007; Salichos and Rokas, 2013). Maximum likelihood analysis of plastomes resulted in a resolved and well-supported phylogeny with a topology in conflict with that of the species tree, especially among temperate North American species (Fig. 2, right). Relationships in this clade estimated from noncoding plastid sequences have been shown to be at odds with expectations based on morphology (Fishbein et al., 2011).
CONCLUSIONS
Hyb-Seq, the combined target enrichment and genome skimming approach presented here, efficiently generates copious data from both the low-copy nuclear genome and high-copy elements (e.g., organellar genomes) appropriate for phylogenomic analyses in plants. With a small investment to generate a genome and transcriptome for an exemplar or the utilization of quickly growing resources from the many publicly available genome and transcriptome projects, a probe set can be designed that will target conserved regions that are phylogenetically informative across plant genera or families. Because this approach recovers sequences that are hundreds of base pairs in length from hundreds to thousands of loci, even with modest levels of variation the data are appropriate for addressing questions at low taxonomic levels. Furthermore, sequences flanking the conserved target regions will generally evolve more rapidly, providing additional potentially informative variation.
Fig. 1.
Histogram of exon sequence divergence between the species used for probe design, Asclepias syriaca, and four other species: the most divergent species of Asclepias, A. flava; another member of Asclepiadinae (Asclepiadeae: Asclepiadoideae), Calotropis procera; a member of Gonolobinae (Asclepiadeae: Asclepiadoideae), Matelea cynanchoides; and a member of a different subfamily, Catharanthus roseus (Rauvolfioideae). Note that a maximum sequence divergence of 75% was allowed for BLAT and that exons with >10% divergence were less likely to be observed in Calotropis and Matelea because they were less likely to be enriched by the probes, while the Catharanthus data were from transcriptome sequences of multiple tissues and not subject to target enrichment bias.

Table 3.
Number of single-copy genes recovered for phylogenomic analysis with different data analysis pipelines.

The Hyb-Seq protocol based on taxon-specific genome and transcriptome data has advantages over alternative approaches, such as transcriptome sequencing or genome reduction via restriction digest. Transcriptome sequencing results in thousands of orthologous nuclear loci, but requires living, flash frozen, or specially preserved tissue for RNA extraction, is subject to large amounts of missing loci across samples, and does not as effectively sample rapidly evolving noncoding regions. In contrast, target capture and genome skimming can use small amounts of relatively degraded DNA, such as extractions from herbarium specimens (Cronn et al., 2012; Straub et al., 2012), and consistently yield intron and 5' and 3' untranslated region sequence. Genome reduction methods utilizing restriction digests (e.g., RAD-Seq, genotyping-by-sequencing; Davey et al., 2011) also produce thousands of loci, and have been effective in resolving phylogenetic relationships and patterns of introgression (e.g., Eaton and Ree, 2013). However, the effectiveness of these approaches with poor quality or degraded DNA has not been demonstrated, and the anonymous nature of these loci makes it more challenging to determine orthology. Most importantly, the data obtained (SNPs or 30–200-bp sequences) are not appropriate for applying phylogenetic approaches that estimate species trees from a large number of gene trees. Focusing on orthologous targets through Hyb-Seq also reduces the amount of missing data relative to both transcriptome and RAD-Seq studies. Until the sequencing of whole genomes for every species of interest becomes practical and affordable, the protocol presented here is poised to become the standard for quickly and efficiently producing genome-scale data sets to best advance our understanding of the evolutionary history of plants.
Fig. 2.
Comparison of the species tree of Asclepias based on 761 putatively single-copy loci and the whole plastome phytogeny. The MP-EST tree is shown at left, and the difference between this topology and that recovered through an analysis of the concatenated nuclear gene data set is indicated by the red arrow. Solid lines connect each species to its placement in the plastome phytogeny (right). Values near the branches are bootstrap support values (* = 100%). Colors reflect the plastid clades of Fishbein et al. (2011): temperate North America (green), unplaced (orange), highland Mexico (purple), series Incarnatae sensu Fishbein (pink), Sonoran Desert (blue), and outgroup (black).

LITERATURE CITED

Notes
[1] The authors thank M. Dasenko, Z. Foster, K. Hansen, S. Jogdeo, Z. Kamvar, L. Mealy, M. Parks, M. Peterson, C. Sullivan, and L. Worchester for laboratory, sequencing, data analysis, and computational support. J. M. Rouillard with MYcroarray provided valuable technical support. The authors thank J. Mandel and another anonymous reviewer for comments on a previous version of this manuscript. This work was funded by the U.S. National Science Foundation (DEB 0919583).