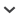Vertebrates are part of the phylum Chordata, itself part of a three-phylum group known as the deuterostomes. Despite extensive phylogenetic analysis of the deuterostome animals, several unresolved relationships remain. These include the relationship between the three deuterostome phyla (chordates, echinoderms and hemichordates), and the monophyletic or paraphyletic origin of the cyclostomes (hagfish and lampreys). Using robust Bayesian statistical analysis of 18S ribosomal DNA, mitochondrial genes and nuclear protein-coding DNA, we find strong support for a hemichordate-echinoderm clade, and for monophyly of the cyclostomes.
INTRODUCTION
Within the animal kingdom (Metazoa), the triploblasts or Bilateria are usually divided into two groups, protostomes and deuterostomes, on the basis of developmental and adult morphological characters. Most invertebrates are protostomes and generally develop the mouth, or the mouth and anus, from the blastopore (protostome = “first mouth”). Deuterostomes generally develop only the anus from the blastopore, the mouth forming as a secondary opening (deuterostome = “second mouth”). There is now a consensus that the deuterostomes comprise just three animal phyla: Chordata (including the vertebrates or craniates), Hemichordata and Echinodermata. Molecular phylogenetic studies have provided additional support for grouping these three phyla, while also removing some additional phyla (e.g. Chaetognatha) from the deuterostomes into the protostomes (Field et al., 1988; Telford and Holland, 1993; Turbeville et al., 1994; Wada and Satoh, 1993). The relationship between the three deuterostome phyla is less clear. Widely varying body plans between these phyla make developmental and morphological comparisons difficult, while molecular evidence from 18S rDNA and mitochondrial DNA has proved suggestive but not conclusive in this regard.
For most of the twentieth century, the accepted relationship between the three deuterostome phyla placed the hemichordates as sister group to the chordates, to the exclusion of the echinoderms. This was originally based upon a putative homology between the notochord of chordates and the stomachord of hemichordates (Bateson, 1884). This putative homology gains no support from gene expression analysis. For example, the Brachyury gene is expressed in the notochord of all chordates analysed but its orthologue is not expressed in the stomachord of hemichor-dates (Peterson et al., 1999). Another notable similarity between chordates and hemichordates is the presence of U-shaped pharyngeal slits. The expression pattern of Pax1/9 class genes suggests that chordate and hemichordate pharyngeal gill slits are likely to be homologous (Ogasawara et al., 1999).
The second possible relationship between the three deuterostome phyla groups echinoderms with hemichor-dates; this clade is referred to as the Ambulacraria (Metschinkoff, 1881). These two phyla share a similar dipleurula larval morphology (Nielsen, 1997) although some have argued that these similarities may have originated by convergent evolution (Nezlin, 2000). Phylogenetic analysis of mitochondrial and 18S DNA sequence data (Castresana et al., 1998a; Cameron et al., 2000) and a combination of the two (Bromham and Degnan, 1999) offered reasonable support for this grouping. Intriguingly, representatives of the two phyla also share a slightly modified mitochondrial genetic code (Castresana et al., 1998a, 1998b).
The third possible grouping, sometimes referred to as the calcichordate grouping, is based on interpretations of unusual fossils known as cornutes and mitrates (Jefferies, 1986; Jefferies et al., 1989). The anatomy of these fossils can be interpreted as showing a mixture of chordate and echinoderm-like characters, with particular fossils being compatible with placement in the stem group for echinoderms plus chordates (Jefferies and Jacobson, 1998). Other authors have disputed these interpretations, classifying cornutes and mitrates as echinoderms (Ubaghs, 1975). No molecular data has provided support for the calcichordate grouping.
Hagfish and lampreys were originally classified into a single taxon known as the Cyclostomata (Dumeril, 1806; Romer, 1966). Later cladistic analyses of morphological and physiological characteristics threw this into doubt, strongly suggesting that lampreys were the sister group of the gnathostomes (Løvtrup, 1977; Forey; 1984; Maisey, 1986). In contrast, analysis of ribosomal DNA and nuclear protein coding sequences have tended to support the original model (monophyly of the cyclostomes) although the level of support has been rather variable (Stock and Whitt, 1992; Mallatt and Sullivan, 1998; Kuraku et al., 1999). Analyses of mitochondrial data undertaken to date has been unable to find support for one grouping over the other (Delabre et al., 2000).
Since phylogenetic ambiguity still remains concerning these nodes, we undertook a molecular phylogenetic analysis of deuterostome animals, reanalysing the 18S rDNA and mitochondrial gene data and also analysing five nuclear protein coding genes (BMP2/4, Brachyury, Otx, SOD, TPI). In an attempt to increase the robustness of the analysis beyond that previously performed, we utilised a Bayesian statistical procedure based on Monte Carlo Markov Chain (MCMC) sampling methods. Such methods have a rigorous statistical basis, allowing the confidence in a particular hypothesis to be assessed. In effect, these methods calculate the probability that a specified phylogeny is correct, given the model and the data used (Shoemaker et al., 1999). The support given for each topology is directly interpretable simply as a posterior probability, unlike the standard (non-parametric) bootstrap. Such methods have only recently been applied to real molecular phylogenetic questions but have been shown to be extremely powerful (Bollbank and Heulsenbeck, 2001; Lutzoni et al., 2001)
MATERIALS AND METHODS
Phylogenetic analysis
Protein sequences were aligned using ClustalW within the sequence editor package BioEdit (Hill, 1999) and further edited by eye when necessary. In the mitochondrial dataset, each protein was aligned separately and then the alignments concatenated (to avoid alignment problems caused by changes to mtDNA gene order). Concatenation followed the method of Bromham et al. (1998) and aimed to include only sites which are informative over deep divergences. For both nuclear and mtDNA protein-coding sequences, alignment was carried out on the inferred amino acid sequence; this was used as the template to align the original nucleotide sequences, which were used for further analysis. Third codon positions were excluded from analysis to reduce problems caused by substitution saturation. The 18S rDNA sequences were aligned following the alignment used by Bromham and Degnan (1999). BAMBE version 1.01 Beta (Simon and Larget, 2000) with the HKY substitution matrix and gamma rate heterogeneity was used for all tree-inference. This was used exactly as described in the online manual with several preliminary runs carried out to fine-tune the parameters used. In each run, at least 500,000 cycles were performed, starting from a random tree. A burn-in of at least 10,000 cycles was allowed, followed by tree-sampling every hundredth cycle. The posterior probability of a given topology is proportional to the number of times the program visits that particular topology, assuming the Markov chain to have reached stationarity. Stationarity is estimated to have occurred when the topologies sampled reach and maintain an optimum probability, which can be assessed by plotting the probability output against cycle number. All data outside this “plateau” region were discarded. In all cases, the probability plots were also examined to check that convergence had not occurred. Convergence manifests in a sine wave shape when the plateau region is graphed, and was never seen in this study (data not shown). The sampling from the plateau region of the output usually yielded 4500 or more trees, which were then analysed using the program Summarize within the BAMBE package. Obtaining consistent results from several different starting points is a criterion for confidence; hence, five runs were carried out for each dataset starting from a random tree and generating a different random number set each time. In every dataset, each run produced the same most probable tree topology with similar posterior probabilities for each node. The most probable topology was then used to generate maximum likelihood branch lengths using Tree-Puzzle version 5.0 (Strimmer and von Haeseler, 1996), and a mean was taken of the posterior probabilities of each node to provide a consensus tree. Posterior probabilities were attached to each alternative hypothesis by counting the number of trees which corresponded to each topology over all five runs.
Data
The three types of gene analysed (mtDNA, nuclear rDNA and nuclear protein-coding genes) have different modes of molecular evolution, with regard to relative propensity of indels versus substitutions, rates of evolution, and mode of inheritance. For this reason, we elected to analyse the three types of data independently, and not to merge different types of data. All datasets included several chordates and at least one hemichordate, echinoderm, and protostome outgroup species. In many cases, sequences from urochordates and cephalochordates were also available. However, the MCMC analyses revealed these to be very unstable within each phylogeny, thereby increasing the number of different trees retrieved and decreasing support values for all nodes. We therefore removed these basal chordates from the analyses. Smaller datasets were also used to reduce computation time, especially since several runs were carried out for all datasets. The following DNA sequences were retrieved from GenBank: 18S rDNA: Xenopus ( X04025), Sebastolobus ( M91182), Petromyzon ( M97575), Eptatretus ( M97572), Myxine ( M97574), Saccoglossus ( AF236801), Balanoglossus ( AF278685), Asterias ( M20114-6), Arbacia ( M20050-2), Argopecten ( L11265), Spisula ( L11271), Drosophila ( M20062-4). Mitochondrial DNA: Homo ( AB055387), Danio ( AC0-24175), Xenopus ( M10217), Lampetra ( Y18683), Myxine ( AJ404-477), Balanoglossus ( AF051097), Strongylocentrotus ( X12631), Arbacia ( X80396), Locusta ( X80245), Albinaria ( X83390). BMP2/4: Xenopus BMP2 ( X55031) BMP4 ( X63426), Danio BMP2 ( U82233) BMP4 ( U82231), Lytechinus ( AF119712), Ptychodera ( AB028219), Drosophila ( M30116). Brachyury: Homo ( NM_009209), Mus ( NM_-003181), Gallus ( U25176), Asterina ( AB018527), Hemicentrotus ( D50332), Ptychodera ( AB004912), Platynereis ( AJ289022). Otx: Homo ( NM_021728), Rattus ( NM_013109), Xenopus ( Z46972), Danio Otx1 ( D26172), Otx2 ( D26173), Otx3 ( D26174), Ptychodera ( AB028220), Strongylocentrotus ( S76899), Tribolium ( AJ223627), Drosophila ( X58983). TPI: Homo ( NM_000365), Mus ( NM_009415), Gallus ( M11941), Lampetra ( AB025327), Eptatretus ( AB025322), Branchiostoma ( AB000892), Anopheles ( U82707), Drosophila ( AF025814). SOD: Homo ( NM_000636), Rattus ( NM_017051), Gallus ( AF329270), Petromyzon ( X64059), Eptatretus ( X64058), Bran-chiostoma ( X64061), Parastichopus ( X64060).
RESULTS AND DISCUSSION
Selection of genes
18s rDNA and mitochondrial DNA sequence data have been used previously for analysis of deuterostome relationships e.g. (Castresana et al., 1998a; Cameron et al., 2000; Bromham and Degnan, 1999; Stock and Whitt, 1992; Mallatt and Sullivan, 1998; Delabre et al., 2000). In general, these have pointed towards monophyly of the Ambulacraria and (with weaker support) monophyly of cyclostomes. Support values for these groupings have varied between analyses, however, in some cases being very low. Furthermore, previous analyses have used bootstrapping or maximum likelihood support values as indicators of tree robustness, and these are difficult to relate precisely to statistical probability values. We reasoned, therefore, that it would be informative to re-analyse 18S rDNA and mtDNA data using newly developed and powerful Bayesian statistical methods. Furthermore, this would provide a useful test case to assess the power of this phylogenetic analysis method. Nuclear protein-coding genes have been used more rarely to investigate deuterostome relationships. We suggest that these are also worthy of analysis using Bayesian statistical methods, thereby affording the opportunity to compare and contrast results obtained form three independent sets of genetic information. Our choice of nuclear-protein coding genes was limited by relative scarcity of sequence data for hagfish and hemichordates. Indeed, no suitable genes had been sequenced in all deuterostome phyla and in both hagfish and lamprey. Database and literature searches yielded three protein-coding genes suitable for analysis of echinoderm and hemichordate relationships: the Otx class homeobox genes, the Brachyury gene (both these encode transcription factors) and the Bone Morphogenetic Protein (BMP)-2/4 genes (encoding secreted signalling molecules). Two protein-coding genes were suitable for analysis of cyclostome relationships; these encode the enzymes manganese-type superoxide dismutase (SOD) and triose phosphate isomerase (TPI).
Deuterostome phylogeny
Deuterostome phylogeny was investigated using nuclear 18S rDNA, concatenated mitochondrial protein-coding genes, and three nuclear protein-coding genes (BMP2/4, Otx, Brachyury). All five datasets support the same most probable tree-topology (Fig. 1A–E). This topology groups the hemichordates and echinoderms together to the exclusion of the chordates. Posterior probability support (the probability that this clade exists, given the model and the data) for the hemichordate-echinoderm clade varied from 1.000 to 0.958. The mean posterior probability over the five datasets was 0.990. Given that five different datasets, from three (semi-) independent parts of the genome, were used this is very strong support for the existence of a hemichor-date-echinoderm clade.
Fig. 1
A-G: Most probable tree-topologies generated by Bayesian statistical analysis, showing maximum likelihood branch lengths. Support for each node is given by posterior probabilities. Animals are identified by genus. Support for the Ambulacraria clade is indicated by a filled arrow. Support for the monophyly of the cyclostomes is indicated by a white arrow. H: Summary of conclusions from these analyses.

A second way of analysing the output is to compare the probabilities of the three alternative biologically plausible topologies, simply by counting their frequency within each analysis output. We find the most probable of the three trees is always the Ambulacraria hypothesis (tree A). The probability of the traditional topology (T) and the calcichordate topology (C) are comparable to each other, and very low (Table 1). This method of interpreting the output is more stringent than probability calculation for each node of the “most probable” tree, because (for instance) a tree which contains an hemichordate-echinoderm clade is only classified as supporting Ambulacraria (A) when this group also lies as a sister group to a monophyletic Chordata. For the 18S rDNA and mtDNA analyses, every tree recovered matched the Ambulacraria hypothesis perfectly (out of more than 20,000 trees sampled). The paraphyly effect was seen infrequently, however, on analysis of nuclear protein-coding genes. For example, almost all BMP2/4 trees placed the echinoderms and hemichordates as a monophyletic group (99.2%), but in a small number of these cases the clade was placed within the chordates. Therefore the posterior probability of a hemichordate-echinoderm clade (0.992) is higher than the probability of the Ambulacraria tree (0.815). This phenomenon is even more marked for Brachyury and Otx. These two genes encode transcription factors, which are subject to strong selection in the DNA binding domains; this may make them more problematic for phylogenetic analysis. In these two data sets, a high proportion of trees (>40%) do not match one of the three alternative hypotheses. Again, this was usually caused by a hemichordate-echinoderm clade falling within the chordate group instead of being a sister group to it. This problem could be reduced under a Bayesian framework, by assigning low prior probabilities to “biologically implausible” trees; we chose not to do this, treating all prior probabilities as equal. Even under our highly stringent parameters, however, the Ambulacraria tree always gained vastly higher statistical support than either the calcichordate tree or the traditional tree, in every data set examined.
Table 1
Posterior probabilities of the alternative topologies (T, A or C), inferred by counting the number of trees with each topology in the sampled dataset. T = Traditional, A = Ambulacraria (Echinoderm-Hemichordate), C = Calcichordate.

We conclude that the hemichordates and echinoderms form a natural group (the Ambulacraria), and that this is a sister group to the chordates. There are important biological implications of this phylogeny. First, those characters used traditionally to unite hemichordates and chordates must be re-evaluated. The most important character is undoubtedly the presence of pharyngeal slits formed by fusion of endoderm and ectoderm, seen in hemichordates (both enteropneust and pterobranchs) and in chordates (urochordates, cephalochordates and vertebrates). Recently gene expression analyses have confirmed that these structures are probably homologous, since both enteropneust and chordates express Pax-1/9 genes during formation of the pharyngeal slits (Ogasawara et al. 1999). We conclude that living echinoderms have lost pharyngeal slits during evolution. In this context it is noteworthy that some enigmatic fossils known as carpoids have probable pharyngeal slits, but also a calcite skeleton strongly indicative of echinoderm affinity (Jefferies 1986). There is controversy surrounding the precise phylogenetic position of these diverse animals, but at least some are likely to be stem group echinoderms. If correct, the presence of slits in carpoids adds weight to our contention that living echinoderms have lost pharyngeal slits. Other putative shared anatomical characters between chordates and hemichordates, such as the notochord/stomachord and a postanal tail are less significant; the former is a dubious homology with little embryological support and no gene expression similarity (Peterson et al., 1999), while the latter may be an example of convergent evolution. The second point to consider is whether hemichordates and echinoderms have any shared anatomical or embryological characters that evolved on their common stem lineage. The best candidate is the nature of their larva; indeed, the remarkable similarity of some echinoderm larvae and hemichordate tornaria larvae has been noted for over 100 years (Garstang, 1894); our analyses serve to strengthen the case for these being homologous larval forms.
Cyclostome monophyly
Cyclostome phylogeny was investigated using nuclear 18S rDNA, concatenated mitochondrial protein-coding genes, and two nuclear protein-coding genes (SOD, TPI). All four datasets supported the same most probable tree topology, including a monophyletic grouping of lampreys and hagfish (Fig. 1, A, B, F, G). The posterior probabilities supporting this clade were extremely high for three data sets (1.000 mtDNA; 0.995 18S rDNA; 0.946 TPI), but lower for the fourth (0.761 SOD). The mean posterior probability for the monophyly of the cyclostomes over the four datasets was 0.926. We note in particular that the Bayesian analysis of the mitochondrial dataset performed here yielded very strong support for the cyclostome clade, when other methods of phylogenetic reconstruction provided only tentative support using similar input data (Delabre et al., 2000).
As with the deuterostome analysis above, we also compared the statistical support for the two biologically plausible hypotheses (cyclostome monophyly versus lampreys as sister to gnathostomes). The probability of the monophyletic grouping was found to be considerably higher than the probability of the paraphyletic grouping in all cases (Table 1). We conclude that the lampreys and hagfish form a natural group (the cyclostomes). There are some important biological implications of this phylogeny. First, consider the anatomical and physiological characters that are shared between lampreys and jawed vertebrates, but not seen in hagfish. These include the presence of true vertebrae with well developed neural and haemal raches, radial muscles in the fins, a lateral line with neuromast organs, extrinsic eye muscle and hyperosmoregulation (Forey and Janvier, 1993). Under the molecular phylogeny reported here, we conclude either that these characters evolved convergently in lampreys and jawed vertebrates, or they have been lost in hagfish. The latter seems more likely, suggesting that the hagfish body plan has become secondarily reduced in anatomical complexity during evolution. Second, there are characters that are uniquely shared by lampreys and hagfish, including a lack of dermal armour, presence of a single median nostril, a circular mouth and pouch-like gills (Forey and Janvier, 1993). We conclude that some or all of these characters are homologous between lampreys and hagfish. Third, there is a taxonomic implication. It has become common practice amongst systematists to reserve the term “vertebrate” for a clade containing lampreys and jawed vertebrates, to the exclusion of hagfish (the latter being invertebrate craniates). The phylogenetic tree favoured here rejects this terminology, because there is no clade uniting lampreys and jawed vertebrates. Consequently, we consider craniates and vertebrate to be equivalent taxonomic terms, and favour retaining the well known term “vertebrate” for lampreys, hagfish and jawed vertebrates.
Table 2
Posterior probabilities of the two alternative cyclostome topologies (monophyly or paraphyly), inferred by counting the number of trees with each topology in the sampled dataset.

The power of Bayesian analysis for phylogenetics
We found that the Monte Carlo Markov Chain method for phylogenetic analysis of DNA sequences, as implemented in BAMBE, produced consistent results from three types of data set with rather different modes of evolution. Furthermore, the results we obtained strongly indicated the existence of two distinct phylogenetic groupings (cyclostomes and Ambulacraria) that had been proposed previously from molecular data; our results are consistent with previous methods in this respect. The support values obtained from BAMBE were consistently higher than those produced by other methods. We interpret this to mean that BAMBE has recovered the biologically correct phylogenetic tree with greater certainty than did previous methods. In effect, the existence of a monophyletic Ambulacraria and monophyletic cyclostomes is elevated from being merely plausible to being extremely likely. The concurrence between data sets gives us additional confidence in these results, although we caution that the susceptibility of MCMC methods to problems such as long-branch attraction has not yet been assessed
From a theoretical standpoint, a clear advantage of these methods is that support for particular clades can be interpreted directly as a probability. Confusion over the statistical ‘meaning’ of the bootstrap value is therefore avoided. Using this method with 18S rDNA and mitochondrial DNA, monophyly of the cyclostomes and existence of an hemi-chordate-echinoderm clade are both supported with greater than 99% statistical support. It is also possible to determine the probability of a given topology, and compare it to several alternatives, as well as to infer the strength of phylogenetic signal in the data from the number of biologically implausible trees obtained. For example, our trees drawn from nuclear protein-coding sequences concurred with those drawn from 18S rDNA and mtDNA, but the statistical support for key nodes was often lower. This was usually due to a high frequency of implausible trees (rather than high incidence of the biologically plausible alternative topologies), indicating weakness, but not absence, of phylogenetic signal in individual protein-coding genes.
From both empirical and theoretical standpoints, therefore, we argue that Bayesian statistical procedures based on Monte Carlo Markov Chain (MCMC) sampling methods comprise an extremely powerful set of tools for molecular phylogenetic inference.
Acknowledgments
We thank Mark Pagel for suggesting use of Bayesian statistical method for these analyses, and for his help with using BAMBE. We also thank two anonymous referees for their comments. This project was funded by the BBSRC.