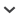The genomes of vertebrates are globally methylated, but a small portion of genomic regions are known to be hypomethylated. Although hypomethylated domains (HMDs) have been implicated in transcriptional regulation in various ways, how a HMD is determined in a particular genomic region remains elusive. To search for DNA motifs essential for the formation of HMDs, we performed the genome-wide comparative analysis of genome and DNA methylation patterns of the two medaka inbred lines, Hd-rRII1 and HNI-II, which are derived from northern and southern subpopulations of Japan and exhibit high levels of genetic variations (SNP, ∼ 3%). We successfully mapped > 70% of HMDs in both genomes and found that the majority of those mapped HMDs are conserved between the two lines (common HMDs). Unexpectedly, the average genetic variations are similar in the common HMD and other genome regions. However, we identified short well-conserved motifs that are specifically enriched in HMDs, suggesting that they may play roles in the establishment of HMDs in the medaka genome.
INTRODUCTION
Methylation of cytosine at CpG dinucleotides is one of the most fundamental epigenetic modifications of vertebrate genomes. DNA methylation is often described as a ‘silencing’ epigenetic mark, as DNA methylation at gene promoters is associated with stable repression of gene expression (Bird, 2002). In vertebrates, a small portion of genomic regions are known to be hypomethylated, and such hypomethylated domains (HMDs) are often seen in gene promoters(Hendrich and Tweedie, 2003). Most of those HMDs serve as a site for binding of transcription factors and accumulate histone modifications, mostly active but sometimes of repressive-type (Jeong et al., 2014; Nakamura et al., 2014), and thereby contribute to transcriptional regulation of nearby genes. In addition to these promoter-associated HMDs, some HMDs are seen in regions distant from promoters. Recent studies have reported a wide variety of functions of DNA methylation at gene bodies and inter-genic regions, such as regulation of splicing, alternative promoters, enhancers, and insulators (Jones, 2012). Hence, the questionof how HMDs are established and, in particular, how an HMD is determined in a particular genomic region, has been a subject of intense studies in genome science.
Cis-regulatory sequences are thought to initially determine the epigenetic code, a combination of DNA methylation and histone modifications. Indeed, the analysis using hybrid mice of two inbred lines demonstrated that DNA methylation patterns are regulated by cis-sequences (Schilling et al., 2009). Consistent with this, a transgenic approach has revealed that the methylation patterns of inserted DNA sequences maintained their original status (Lienert et al., 2011). The strong association between genotype and DNA methylation in human family also suggests the importance of cis-elements (Gertz et al., 2011). However, consensus DNA sequences that regulate the pattern of DNA methylation remain elusive. A simple approach for looking for such cis-elements is the identification of evolutionary conserved genomic sequences among closely related species and relating them to the epigenetic code. Recent advances in DNA sequencing technology have facilitated this approach (Heinz et al., 2013; Kasowski et al., 2013; McVicker et al., 2013). However, it remains difficult to identify conserved motifs even in human and mouse which have rich genome and epigenome resources, because of the low frequency of genetic variations within populations (∼ 0.1%).
In this context, the medaka is a particularly useful model system with the high quality draft genome (Kasahara et al., 2007) and base-resolution methylome (Qu et al., 2012). Importantly, the medaka has polymorphic inbred lines from two geographically separated subpopulations living in the northern and southern part of Japan. The two subpopulations were separated by an appropriate evolutionary distance (4–18 million years) that is close enough to reliably align noncoding sequences but also entails sufficient sequence variations (SNP, ∼ 3%) (Takehana et al., 2003; Kasahara et al., 2007; Setiamarga et al., 2009; for review, see Takeda and Shimada, 2010). The two subpopulations were originally considered as one species, Oryzias latipes, but recently northern one was described as a new species, Oryzias sakaizumii (Asai et al., 2011). However, the two species are biologically similar to each other; they can mate and produce healthy offspring under laboratory conditions, even showing hybrid vigor. Thus, the transcriptional and epigenetic profiles of the two species might be largely conserved under such large genetic variations. Thus, the comparison of the two genomes and methylomes may provide insights into mechanisms of HMD formation mediated by cis-elements.
Here, we performed the genome-wide comparison of genome and DNA methylation patterns of the two medaka inbred lines, Hd-rRII1 and HNI-II, from southern and northern species, respectively. We focused on the genome of blastula in which all cells retain pluripotency, and the epigenome of this stage is so called ‘ground-state’. In the aligned genome regions of the two species, the majority of HMDs were found to be conserved between the two species (common HMDs). Unexpectedly, common HMDs still accumulate genetic variations at a comparable level to that of the methylated regions (∼ 2.8%). However, we identified short well-conserved motifs that are enriched in HMDs.
MATERIALS AND METHODS
Fish strains
We used medaka Hd-rRII1 (referred to as Hd-rR), d-rR, and HNI-II (referred to as HNI). Medaka fishes were maintained and raised under standard condition. All experimental procedures and animal care were carried out according to the animal ethics committee of the University of Tokyo.
Identification of common HMDs and species-specific HMDs
First, we mapped the bisulfite-treated reads collected from HNI blastula-stage embryos (Qu et al., 2012) to the HNI genome (version 2) which became available recently ( http://mlab.cb.k.u-tokyo.ac.jp/~yoshimura/Medaka/#!Assembly.md), using the mapping conditions previously described (Qu et al., 2012). We then identified HMDs in HNI blastula embryos based on the same criteria as our previous report (Nakamura et al., 2014).
Next, we mapped the identified HMD sequences of each species to the genome of the other species using BLAT (tileSize = 18, oneOff = 1) (Kent, 2002), as the mapping with such parameters are compatible with both high sensitivity (> 99.9% are expected, data not shown) and short calculation time. Among the outputs, due to partial similarities, queries were sometimes mapped to much longer genomic regions. A majority of such cases seemed to represent mapping errors, because insertion or deletion events of > 2 kb regions were rare in the regions which were reliably aligned between the two species. Thus, in order to obtain reliable comparison, we did not include the outputs for further analysis in which the mapped region's length is > 2 kb longer than that of query HMD. After removing these outputs, we further isolated query sequences (hypomethylated sequences in Hd-rR or HNI) which were uniquely mapped or multiply mapped to other genomic regions. We set a criterion that 80% of query's sequences were aligned in the other species' genome. This criterion excluded 1% (Hd-rR mapped to HNI) or 4% (HNI to Hd-rR) of uniquely mapped pairs and 92% (both cases) of multiple mapped pairs. To further isolate reliable pairs from the remaining multiply mapped outputs, we extracted pair as reliable ones of which the best matching rate of such pair (the ratio of the number of the base matches to the whole query size) was > 50% higher than that of any other pairing.
Subsequently, from the remaining results, we selected those in which the mapped genomic region of the query HMD was unique and was not covered by any other query HMDs. Last, we extracted the mapping results in which the query HMD was anchored to the same chromosome.
For the remaining results, we checked if each HMD of the target genome overlapped with the mapped region of the query HMD. If the test was negative, we regarded that such an HMD had no corresponding HMD in the other species and identified it as a ‘species-specific HMD’; otherwise, we treated it as a ‘common HMD’ that is shared in common in both species. Since > 94% of the common HMDs which were identified from the mapping of Hd-rR HMDs to HNI genome overlapped with the common HMDs which were identified from the mapping of HNI HMDs to Hd-rR genome, we used the former set as ‘common HMDs’ in all analyses.
RNA-seq
For d-rR blastula cells, the previously obtained data was used (Nakatani et al., 2015). For HNI blastula cells, RNA was isolated using ISOGEN (Nippon Gene) and RNeasy mini kit (QIAGEN) and treated with Ribominus eukaryote kit for RNA-seq (Life Technologies).RNA-seq library was prepared using TruSeq RNA-seq sample prep kit (Illumina). The PCR products were purified and size fractionated using a bead-mediated method (AMPure, Ambion). Sequencing was conducted on HiSeq 2500 platform (Illumina). Sequences were mapped using BWA (Burrows-Wheeler Alignment tool) (Li and Durbin, 2009) and RPKM (reads per kilobase of exon per million mapped reads) was calculated using SAMMATE software (Xu et al., 2011).
Calculation of the incidence of genetic variations between Hd-rR and HNI
We categorized the HMD sequences into three HMD groups, ‘common HMDs’, ‘Hd-rR specific HMDs’ and ‘HNI specific HMDs’ and similarly classified the corresponding regions on the other species' genome, and performed the alignment of reciprocally best matching pairs of sequences with LASTZ (Harris, 2007) (--format = axt) for each group. As the LASTZ sometimes produced multiple outputs with different size for the same region or outputs that partially overlapped, we removed the relatively short outputs such that the whole aligned region of the query was covered by the longest or second-longest alignments for the same query, then extracted the alignments that were independent and did not overlap with each other. The sequences of gene exons were also excluded from the further analyses. Then, from the remaining output alignments, we counted the single nucleotide polymorphisms (SNPs), insertions and deletions. As a small portion of the mapped regions of common HMDs was methylated in HNI genome (∼ 10% of all mapped regions), we excluded such methylated regions from further analysis of common HMDs. When the alignment of one HMD was separated into more than one block, the mutations of the separated alignments were summed. The mutation rate for each HMD was calculated by dividing the total number of mutations by the length (bp) of the investigated region. For the negative control data set, the original Hd-rR HMD genome-coordinate set was randomly distributed on methylated regions using bedtools (ver. 2.17.0), (Quinlan and Hall, 2010). Then, the obtained sequences of the methylated regions were treated as well as HMDs and used for the calculation of the incidence of genetic variations.
Calculation of 6-mer's mutation index
Using the output of LASTZ alignment, we examined whether a 6-mer is mutated or not by searching the query and the aligned regions for the 6-mer. To take into account the case that short indels occur within a 6 bp aligned region, but 6-mer is still conserved between two medaka genomes in spite of such indels, we extracted the aligned regions flanked by the 8 bp with no-mismatch, and examined whether the 6-mer is conserved within the extracted regions. If the 6-mer in the query was not found in the aligned region, the 6-mer was regarded as ‘mutated’. Then, the mutation index was calculated by dividing the number of ‘mutated’ 6-mers by the total number of the 6-mers in the query. The calculation results of the motif and its reverse complement were combined for each 6-mer.
Motif analyses
TOMTOM (Gupta et al., 2007) was used to search motifs similar to top 20 selected 6-mers. JASPAR Vertebrates and UniPROBE Mouse databases were used as target motifs. We set the significance threshold (q value < 0.1) in the selection of outputs.
Data access
All sequence data are deposited at the NCBI Sequence Read Archive (SRA) ( http://www.ncbi.nlm.nih.gov/sra) (accession number SRP070096).
RESULTS
HMDs are highly-conserved in the closely related medaka species
We first calculated the proportion of HMDs shared by the two inbred lines, Hd-rR and HNI. We previously reported 15,145 HMDs containing at least 10 continuous low-methylated (methylation rate < 0.4) CpGs in Hd-rR blastula embryos (Nakamura et al., 2014). Based on the same criteria, we identified 16,361 HMDs in the HNI blastula embryos using the previously obtained bisulfite-sequencing data (Qu et al., 2012) and a newly assembled genome of HNI (available from http://mlab.cb.k.u-tokyo.ac.jp/~yoshimura/Medaka/#!Assembly.md). We mapped HMD sequences in one species' to the other species' genome and checked if the HMDs are shared by the two species (Fig. 1A). Due to repetitive sequences and deletions (or insertions), about 13% and 23% of Hd-rR and HNI HMDs, respectively, did not map to the other genome. Of the uniquely mapped HMDs (13,165 in Hd-rR, 12,660 in HNI), approximately 95% (that is, ∼ 83% of total Hd-rR HMDs, ∼ 74% of the total HNI HMDs) was commonly found in the two genomes (referred to as ‘common HMDs’) (Fig. 1B, C). Only small populations (618 or 598 HMDs in Hd-rR or HNI, respectively) had no corresponding HMDs in the other species' genome (referred to as ‘species-specific HMDs’), even though the sequences were uniquely mapped in both genomes. The size of these species-specific HMDs was relatively small compared to that of the common HMDs (see Supplementary Figure S1 online (10.2108.zsj.33.358.s1.pdf)).
As the HMD generally overlaps with the gene promoter (Nakamura et al., 2014), we examined if such tendency is also the case for each sets of HMDs. We found that a large part of the common HMDs (76.1%) are located at promoter regions; 5 kb upstream to 2 kb downstream of transcription start sites (TSSs). On the other hand, fewer than one-third of species-specific HMDs were at gene promoters (29.4% for Hd-rR specific and 27.9% for HNI) (Fig. 1D). Instead, about half and about one-fifth of of the species-specific HMDs were found in regions outside genes and gene bodies (both exon and intron), respectively.
Species-specific HMDs affect gene transcription
We examined how each type of HMDs (common or species-specific) is reflected in gene transcription of the two species, by conducting a comparison of RNA-seq data. We newly obtained about 62.5 M reads from HNI blastula cells, and for Hd-rR, we utilized our previous RNA-seq data from d-rR (Nakatani et al., 2015), a closed colony line from which the Hd-rR inbred line had been established. After mapping them to the Hd-rR genome, genes were isolated and classified according to those having common HMDs or species-specific HMDs in their promoter regions or in gene bodies. As for the HMDs located in inter-genic regions, we searched for their nearest genes. In order to compare the relative expression level, we calculated the ratio of RPKM (reads per kilobase of exon per million mapped sequence reads) , the gene expression level normalized by the total number of the mapped reads and the length of exon, of d-rR to that of HNI (d-rR/HNI) for each gene. In the genes with common HMDs in their promoters, the median of the RPKM ratio was 0.86 (Fig. 1E, left, green), which deviate a little from the ideal figure, 1.0, probably due to slightly different conditions (e.g. sampling timing and experimental procedures) in the two independent RNA-seq experiments. In spite of this, we found a significant tendency; the expression ratio of the genes with Hd-rR-specific HMDs and HNI-specific HMDs in their promoters was significantly higher (0.97) and lower (0.60) than those with common HMDs in their promoters, respectively (Fig. 1E, left, pink and blue). This suggests that the genes of which promoters are marked by HMDs tend to express at higher levels than their unmarked counterparts. The expression level of the two species in each gene which has a species-specific HMD in the promoter is provided in Supplementary Table S1 and S2 (10.2108.zsj.33.358.s1.pdf) (Hd-rR specific in S1, HNI specific in S2). In genes which have a HMD in gene bodies or inter-genic regions, the ratio of RPKM did not significantly change between each gene category (Fig. 1E, middle and right).
Genetic variations between Hd-rR and HNI in HMDs
High conservation of HMDs in the two divergent genomes could be explained if genetic mutation occurs less frequently in those HMD regions. To test this idea, we investigated the rate of sequence variations within the common HMDs, species-specific HMDs and methylated regions. Unexpectedly, however, the average frequency of polymorphisms (SNPs) did not show a significant difference among those regions; the median was 2.77, 2.75, 2.83 and 2.96% for methylated, common, Hd-rR-specific and HNI-specific, respectively (Fig. 2, left). Thus, the incidence of genetic variations in common HMDs is comparable to that in the methylated regions.
In contrast with SNP, the indel (insertion/deletion) rate was higher in the common HMDs (Fig. 2, right). This may be due to the fact that HMDs marking the promoter are open in chromatin structure and more susceptible to insertion/deletionevents than compact methylated regions. Indeed, the indel rate was reported to show peaks in the regions with the low nucleosome-occupancy downstream of TSS (Sasaki et al., 2009). The indel event, however, is far less frequent as compared with SNP (0.64% (indel) vs. 2.75% (SNP) in common HMDs) and does not affect much on the overall mutation rate.
Fig. 1.
HMDs are highly-conserved between medaka species, Hd-rR and HNI. (A) Schematic representation of HMDs in aligned genomic regions. Hd-rR blastula HMDs are mapped to the genome of HNI to identify common HMDs and Hd-rR specific HMDs. We performed the mapping of HNI HMDs to Hd-rR genome for identifying HNI specific HMDs (not shown). (B) Genome browser view showing the example of common HMDs, Hd-rR specific HMDs and HNI specific HMDs. The distribution of CpG is shown in black vertical lines and the methylation level is shown in orange ones. Black horizontal bars indicate the position of each HMD. The sequences of each HMD and its 2 kb flanking regions were mapped to the other species' genome, and the methylation status of the two species was compared in aligned regions (red-dotted boxes). (C) Venn diagram showing the overlap of HMDs between Hd-rR and HNI. Each picture above the diagram shows male Hd-rR or HNI adult fish. (D) Pie charts showing the proportion of HMD type (promoter (orange), gene body (green) and others (grey)) in common HMDs (left), Hd-rR specific HMDs (middle) and HNI specific HMDs (right). (E) Boxplots showing the ratio of RPKM of d-rR to HNI (d-rR/HNI) of the genes marked by common HMDs (green), Hd-rR specific HMDs (pink) and HNI-specific HMDs (blue). Genes were classified according to the position marked by HMDs, promoters (left), gene body (middle) and promoter-distal (right). In the calculation of the ratio of RPKM, the genes in which RPKM of the either species is 0 are excluded. P-values were calculated using Wilcoxon rank sum test. In the box plots, the bottom and top of the boxes correspond to the 25th and 75th percentiles; the internal band is the 50th percentile (median). The plot whiskers extending outside the boxes correspond to the lowest and highest datum within 1.5 interquartile ranges of the lower and upper quartiles, respectively.

Fig. 2.
Genetic variations between Hd-rR and HNI in HMDs. Boxplots showing the incidence of genetic variations between Hd-rR and HNI. The left figure shows the rate of single nucleotide polymor-phisms (SNPs) per base pair, and the right one shows the rate of insertions and deletions per base pair in the methylated regions (gray), common HMDs (green), Hd-rR specific HMDs (pink) and HNI specific HMDs (blue). Note that exons in the Hd-rR genome and their aligned regions in HNI genome were excluded in this analysis, because the proportions of those regions could vary among the investi-gated HMD set. P-values were calculated using Wilcoxon rank sum test. In the box plots, the bottom and top of the boxes correspond to the 25th and 75th percentiles; the internal band is the 50th percentile (median). The plot whiskers extending outside the boxes correspond to the lowest and highest datum within 1.5 interquartile ranges of the lower and upper quartiles, respectively.

Taken together, blastula-stage HMDs are well-conservedbetween the two medaka species in spite of high incidence of genetic variations.
Specific DNA motifs are conserved and enriched in common HMDs
The above fact that common HMDs exhibit comparable levels of SNPs led us to speculate the presence of short crucial DNA sequences that are specifically conserved during speciation. To search for such sequences, we examined the occurrence of short oligomers and their conservation between the two species in HMDs or in the methylated regions. For each of the 2,080 sequences of 6-bp long DNA oligomers (6-mers), we calculated their occurrence and mutation index (the proportion of mutated to all found 6-mers) in each region.
Given that HMDs are predominantly found at gene promoters, certain DNA motifs could be enriched simply because they are required for gene transcription, but irrelevant to DNA methylation state. To efficiently extract the candidate 6-mers essential for HMD patterning, we looked at their mutation index in species-specific HMDs where 6-mers relevant to HMD patterning were expected to be normally mutated. For this, we utilized the ratio of the mutation index of common HMDs to that of species-specific HMDs for assessment. The low value of this ratio indicates that 6-mer is preferentially conserved in common HMDs, but not in species-specific HMDs. Furthermore, since CpGs tend to be more conserved within HMDs, as they are easily lost when methylated (Coulondre et al., 1978; Bird, 1980; Shen et al., 1994), we classified all 2,080 6-mers into two categories by the presence of CpG, and compared their ratio separately. As expected, the histograms of oligomers of each category (Fig. 3A) demonstrate that most of the 6-mers with CpGs and about a half of the 6-mers without CpGs are more conserved in common HMDs (the ratio of mutation index is < 1.0). This result further confirmed the higher conservation level of non-methylated CpGs in HMDs. Then, top twenty most conserved 6-mers in common HMDs (the ratio of mutation index is < 0.455 for CpG and < 0.664 for non-CpG) were selected (Fig. 3B) and subject to further analyses. They are specifically conserved in common HMDs, and could play a role in HMD patterning.
We then examined the enrichment level of each DNA motif in common HMDs. The 6-mers with low ratio of mutation index tended to be highly enriched in common HMDs (data not shown). In particular, top twenty 6-mers of both type (CpG and non-CpG) exhibited significantly higher enrichment levels in common HMDs compared to the methylated regions (Fig. 3C) or species-specific HMDs (see Supplementary Figure S2 (10.2108.zsj.33.358.s1.pdf)). These 6-mers are thus specifically enriched in HMDs and at the same time, well protected against genetic mutations. Finally, we examined the distribution pattern of the conserved 6-mers in common HMDs and found that top twenty 6-mers of CpG and non-CpG are highly accumulated in the HMD region (Fig. 3D).
DISCUSSION
The initial pattern of HMDs examined in this study has a profound effect on gene expression throughout life. Although some methylated genes are later activated by demethylation at their promoters in a cell-type specific manner, the majority of HMDs in blastula cells are largely maintained during development and growth (Potok et al., 2013; Lee et al., 2015).
The medaka system has provided a unique tool to gain insights into genome evolution and speciation (Takeda and Shimada, 2010). In this study, we performed the comparative analyses of genome, expression profile and DNA methylome of the two closely related medaka species, and successfully identified the candidate DNA motifs that may participate in the patterning of HMDs. The estimated divergence time of the two regional species varies depending on a method of estimation; 4–5 million years ago by a molecular clock hypothesis (Takehana et al., 2003) and 18 million years ago by a Bayesian model (Setiamarga et al., 2009). In spite of high accumulation of genetic variations during this long separation time, the two populations had long been considered as a single species, Oryzias latipes. In 2011, however, the northern population was described as a new species, Oryzias sakaizumii (Asai et al., 2011), which is still controversial in the medaka community. In any case, their divergent genetic backgrounds with nearly identical biological features allowed us to survey functional cis-elements throughout the genome. Furthermore, the high-quality draft genome of HNI ( http://mlab.cb.k.u-tokyo.ac.jp/~yoshimura/Medaka/#!Assembly.md), recently produced in addition to Hd-rR, greatly facilitated aligning homologous sequences in the two genomes.
Fig. 3.
Specific DNA motifs are conserved and enriched in common HMDs. (A) Histograms showing the distributions of the ratio of mutation index (common HMDs/Hd-rR specific HMDs) for 6-mers with CpGs (left) or without CpGs (right). (B) Lists of top twenty most conserved 6-mers with CpGs (upper) and without CpGs (lower), which have the lowest values in the ratio of mutation index (common HMDs/Hd-rR specific HMDs). (C) Boxplots showing the 6-mer's enrichment levels in common HMDs (the ratio of each 6-mer's frequency per base pair (common HMDs/methylated regions)). Grey boxes represent all 6-mers, while orange and purple boxes represent the top twenty most conserved 6-mers with and without CpGs, respectively. P-values were calculated using Wilcoxon rank sum test. In the box plots, the bottom and top of the boxes correspond to the 25th and 75th percentiles; the internal band is the 50th percentile (median). The plot whiskers extending outside the boxes correspond to the lowest and highest datum within 1.5 interquartile ranges of the lower and upper quartiles, respectively. (D) Distribution pattern of the top twenty most conserved 6-mers with CpGs (orange) or without CpGs (purple) in the 2 kb region around the boundary of the HMDs of which the size is over 2 kb. The boundaries of HMD were defined at the first low-methylated CpG site inside the HMD. X axis shows the length of each position from the HMD boundary. (E) Schematic representation of conserved DNA motifs and genetic variations in common HMDs and the methylated regions.

As expected from their similar biological features, the pattern of HMDs was found to be highly conserved between the two species. However, we identified a small population of the HMDs (∼ 5% of the mapped HMDs of each species) that were only found in one species. We found that the genes of which promoters are marked by species-specific HMDs tend to express at higher levels than their unmarked counterparts (Fig. 1E, left). This result demonstrated that species-specific HMDs in promoter regions could contribute to species-specific gene transcription. This result is consistent with the previous report of human family that allele-specific DNA methylation accounts for differences in gene expression levels between alleles (Gertz et al., 2011). It should be noted that interpretation of the blastula RNA-seq data is complicated by the presence of maternal transcripts, though the maternal expression profile is expected to reflect the initial HMD pattern as the blastula HMDs tend to be largely maintained during development and growth (Potok et al., 2013; Lee et al., 2015).
Interestingly, the majority of the species-specific HMDs mark the gene bodies or inter-genic regions. This is a sharp contrast to the common HMDs which mostly reside at gene promoters. Consistent with our finding, Hernando-Herraez et al. (2015) reported that most of human-specific DMRs (differentially methylated regions) identified by comparison with non-human primates are located in regions distal to TSSs, although they examined differentiated cells, blood cells, with different methods for identification of the targeted regions. DNA methylation in gene bodies or promoter-distal regions is thought to have diverse functions depending on context, such as transcriptional elongation, alternative splicing, control of alternative promoter usage, and alteration of activity of enhancer or insulators (Jones, 2012), and thereby affects gene expression either positively or negatively. Indeed, in our study, species-specific HMDs located outside gene promoters did not show any correlation with the average relative transcription levels. Notably, the comparison between chick inbred lines demonstrated that DMRs responsible for differences in immune response reside in gene bodies as well as promoters (Li et al., 2015). Taken together, although about 13 or 23% of the HMDs are unmapped in each species, the species-specific HMDs most likely confer species-specific morphological and physiological characters in medaka species (Ishikawa et al., 1999; Kimura et al., 2007; Asai et al., 2011; Tsuboko et al., 2014) and thus will be interesting targets for the future study of speciation.
Species-specific HMDs greatly helped in identifying the conserved short sequences in HMDs. These sequences are specifically enriched and evenly distributed in the common HMDs. Furthermore, they have been protected against genetic mutations for 4–18 million years. Importantly, this specific protection is not observed, when they are located outside the HMD. These facts suggest that the identified short sequences play an important role in initial patterning of HMDs in the blastula genome (Fig. 3E). Thus far, many attempts have been made to identify essential sequences for DNA hypomethylation (Brandeis et al., 1994; Macleod et al., 1994; Dickson et al., 2010; Lienert et al., 2011). Recently, computational analyses have addressed how DNA motifs determine epigenetic status (Luu et al., 2013; Whitaker et al., 2015). Our identified sequences only partially overlapped with those reported motifs (data not shown), raising the possibility that essential motifs vary among species or unknown logic works behind these various motifs. Furthermore, while some of our identified sequences partially overlapped with known binding motifs, more than half of them exhibited no similarity with known motifs (see Supplementary Table S3 online (10.2108.zsj.33.358.s1.pdf)). In any case, we believe that further functional studies of the identified motifs will provide insight into molecular mechanisms underlining the establishment of HMDs, an essential process of genome function.
ACKNOWLEDGMENTS
This research was supported by the Core Research for Evolutional Science and Technology (CREST) program of the Japan Science and Technology Agency (JST), and MEXT KAKENHIGrant Number 25291048. We thank Y. Suzuki, H. Sakata and J. Yoshimura for setting and supporting our computing environment for data analysis. We are grateful to Y. Ozawa and I. Fukuda for fish care.