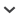It has been believed that clonal propagation by asexual reproduction has serious disadvantages for long-term survival, because asexual reproduction seems not to remove harmful mutations, it seems not to give rise to genetic variations upon which evolution depends and it seems not to reset cell aging. In this article, we re-consider those arguments, by reviewing asexual reproduction of the tunicate, Polyandrocarpa misakiensis. Tracer experiments of bud formation and growth using morphological and chimeric phenotypes showed that the parental epithelial tissues surrounding the bud primordium do not enter the growing bud. It is possible, therefore, to assume that budding involves the purge of a large number of parental somatic cells and tissues. Unlike sexuals, asexuals do not carry out meiotic recombination nor gene shuffling that are two major sources of genetic variation, but we can show that in P. misakiensis at least two genes have significant redundancy and genetic variation even in a clonal colony. Telomerase expressed in germlines is thought to reset the molecular clock executed by telomere shortening. In our Polyandrocarpa cDNA projects, four out of about 2,000 cDNAs examined were matched with retroviral reverse transcriptase that is the catalytic subunit of telomerase, suggesting that telomerase might work in asexual reproduction. In P. misakiensis, dedifferentiation system is used to make new asexual generations. TC14 lectin plays an important role in the maintenance of multipotent but differentiated state of the formative tissue. It is antagonized by tunicate serine protease (TRAMP) that has striking mitogenic and dedifferentiation-inducing activities on the multipotent cells. This system would serve to delay aging of somatic cells. In conclusion, empirical arguments that asexual reproduction is disadvantageous to long-term life do not appear to be tenable to budding of P. misakiensis.
INTRODUCTION
The capacity to repair missing parts of the body is a general characteristic shared by every organism, although the extent of repair varies among species. The regenerative potential of some marine and freshwater invertebrates is so remarkable that a piece of tissues can reconstruct the whole body (Morgan, 1901). Asexual reproduction takes advantage of this potential for propagating individuals (Brien, 1968). It can be found in most phyla in the animal kingdom including Polifera, Cnidaria, Plathelminthes, Annelida, Bryozoa, Echinodermata, Enteropneusta and Chordata.
Asexual reproduction accompanies neither meiotic recombination nor shuffling of male and female genomes, as is usual with sexual reproduction (Barton and Charlesworth, 1998). It is carried out by multipotent somatic cells (cf., Nakauchi, 1982) instead of single germ cells, indicating that the colonial population shares genomic constitution. In the sense that clonal individuals are given off, asexual reproduction has resemblance to parthenogenesis in rotifers (Wurdak and Gilbert, 1977), aphids (Normark, 2000) and others. Actually, evolutional geneticists regard both asexual reproduction and parthenogenesis as synonym (e.g., Wuethrich, 1998). Nonetheless, they are discernible from each other, because parthenogenesis begins with female (sexual) gametes, in which there is a good chance giving rise to genetic recombination and, consequently, genetic variation during oogenesis (Kabay and Gilbert, 1977). In this paper, the term, asexual reproduction, is limited to the narrow definition by which a new individual comes from somatic cells. For broader definition, if necessary, clonal reproduction will be used to refer to the natural creation of clonal individuals.
It has been generally believed that clonal reproduction has disadvantage to long-term survival. This is because clonal reproduction seems not to remove harmful mutations, it seems not to give rise genetic variation and it seems not to reset cell aging. In this paper, we would like to re-consider those classical problems that asexual reproduction addresses, based on our recent findings in the budding tunicate, Polyandrocarpa misakiensis.
Purge of deleterious genes
Many evolutional geneticists argue that the fitness (viability and reproductive success) of sexual reproduction is superior to that of asexual one (Barton and Charlesworth, 1998). They insist that sex serves to assemble beneficial variations and so it creates a well-adapted lineage in the face of a rapidly changing environment (e.g., Ishii et al., 1989). Although genetic mutations are obviously the source of evolution, most mutations affecting fitness appear to be harmful rather than beneficial (Keightley et al., 1998), leading to the extinction of offspring (Fig. 1A). One of the most important merits of sexual reproduction may be, therefore, to eliminate deleterious mutations rather than to accumulate beneficial mutations (Leigh, 1973). Now, we have plenty of hypotheses to explain how sex purges genetic scapegoats (Drake, 1991), although we do not know yet about the accurate rate at which sex removes harmful mutations. Gametogenesis may offer an opportunity to self-diagnose the integrity of genome, by sorting-out of mutations during meiosis (Fig. 1B). Then, harmful genes will be thrown away together with the carrier (gamete) in the trash.
Fig. 1
Fate of harmful mutations in sexual reproduction. (A) In sexual reproduction, when a zygote (top) has harmful mutations (red rod) all descendants have the same deficiencies (middle, bottom). (B) Harmful mutations (red rods) in germlines (top) may be segregated during meiosis (middle), providing a chance to eliminate those mutations in the next generation (bottom).

On the other hand, clonal reproduction seems not to have such an opportunity of sorting-out of harmful mutations. The offspring inherit all of their bad genes and may pick up another through a new mutation (Wuethrich, 1998). In this way, mutations continue to be accumulated in both individuals and in the population (Sniegowski et al., 1997; Taddei et al., 1997). Those notions mentioned above are invariably correct in parthenogenesis where genetic clones are produced by female populations. In the rotifer, Asplanchna sieboldi, diploid females reproduce parthenogenetically via mitotic oogenesis (Gilbert, 1976). This way of reproduction does not give any chances to throw harmful genes away, as a full set of genetic components should be transferred through female generations. Depending on nutritional conditions, some of the females produce eggs undergoing meiotic oogenesis (Kabay and Gilbert, 1997). The haploid eggs develop parthenogenetically into males instead of females. However, these males can scarcely transfer their selected genome to the offspring by fertilization, as the females produce diploid eggs. The accumulation of harmful mutations has a risk leading to the extinction of those populations and, finally, species. As discussed below, asexual reproduction, another mode of clonal reproduction, would not necessarily come into the same consequence as parthenogenesis.
Can asexual reproduction remove harmful mutations?
Tunicates belonging to the phylum Chordata are phylo-genetically the highest organism that can reproduce asexu-ally. Asexual animals of P. misakiensis were first collected in 1970 (Watanabe and Tokioka, 1972), and have ever since been cultured for 30 years in Japanese marine laboratories, including ours. Their mode of propagation is referred to as palleal budding (cf., Nakauchi, 1982). A palleal bud grows out from the parent body wall that consists of the outer and inner epithelia and mesenchymal cells intervening between them (Fig. 2) (Kawamura and Watanabe, 1982a,b; Kawamura and Nakauchi, 1984, 1986).
Fig. 2
Fate of harmful mutations in asexual reproduction. (A) In P. misakiensis, a bud consists of heterogeneous cell populations, the outer epidermal and inner endodermal epithelia and intervening mesenchymal cells. Deficient cells (red boxes) may be extinguished as a result of competition with normal cells (a). If deficient cells are alive but less proliferative than normal cells, they do not have serious effect on the life of clonal individuals (b). When mutant cells have the same or somewhat higher proliferative activity than normal cells, they can occupy a definite area of the animal (c). (B, C) In some cases mutations will be inherited to the next blastogenic generation (right), and in the other cases they will not be inherited (left).

It is noteworthy that the bud begins with heterogeneous cell population instead of a single cell with which sexual reproduction starts. When harmful mutations occur spontaneously in some of those somatic cells, deficient cells may possibly be extinguished as a result of competition with remaining normal cells (Fig. 2A-a). In case they are alive but have less proliferative activity, the probability that they spread to the clonal population becomes very low (Fig. 2A-b). In either cases, harmful mutations would not bring about serious consequence in the clonal life of budding animals. We wonder if deficient cells have similar or somewhat higher proliferative activity than normal cells (Fig. 2A-c). The former (deficient cells) may be substituted gradually for the latter, thus some individuals of the next blastogenic generation being occupied by the offspring of the deficient cells (cf., Figs. 2B and 2C). The spreading (or the block of spreading) of harmful mutations in asexual reproduction depends largely on the amount of parental tissues that participate in bud formation.
In botryllid and polystyelid tunicates, both buds and adult animals are connected with the extra-individual vascular system by orifices (Newberry, 1965; Mukai et al., 1978) (Fig. 3A). In Botryllus, Botrylloides and Symplegma, the orifices are located at definite areas on the zooidal ventral surface. In Polyandrocarpa, on the other hand, they are distributed very abundantly around the basal margin where the bud primordium appears (Fig. 3A), making it easy to trace the epithelial tissue movement during bud growth. Strictly, the orifice is a good landmark of the epidermis but not the inner epithelium. However, many literatures have shown that both epithelia behave synchronously during budding (Berrill, 1941; Izzard, 1973; Kawamura and Nakauchi, 1986). In P. stolonifera, an adult animal of at most 5 mm in length produces very elon-gated buds (stolon) of more than 10 mm in length (Kawamura and Watanabe, 1981). Figure 3B shows the redrawing of their observation of bud growth with reference to orifices. The bud primordium was about 0.3–0.4 mm in width and 0.2 mm in height along the basal margin of the parent body wall. It had two orifices (a, b) at first that were soon divided, respectively, into two sub-orifices (a1, a2, b1, b2). Interestingly, neither a2 nor b2 entered the growing stolon. The orifices increased in distance exclusively between a1 and a2 or between b1 and b2. It is noteworthy that after the bud primordium stage parental epithelial tissues seem not to participate in the stolonial outgrowth.
Fig. 3
Bud growth in P. stolonifera. (A) Adult animal with buds (stolons), ventral view. The test vessel system develops around the basal margin of the body. a, ample; e, endostyle; h, heart; i, intestine; o, orifice; p, pyloric caecum; s, stolon; sti, stigmata; sto, stomach. Bar, 1 mm. (B) Daily growth of a bud. Vascular ampullae are omitted, but orifices (a1, a2, b1, b2) are plotted. As noted by a2 and b2, the parental epidermal tissue does not move toward the growing bud. (Reproduced from Kawamura and Watanabe, 1981).

This empirical observation mentioned above has been supported by chimera experiments (Kawamura and Watanabe, 1984). Two strains of P. misakiensis, white spot and spotless, are discernible from each other by the presence or absence of a white circular spot on the dorsal surface. The color phenotypes are determined by epithelial components of animals (Kawamura and Watanabe, 1984; Ishii et al., 1993). Buds of the spotless strain was amputated and grafted into the body wall of the host adult animal of the white spot strain. The grafts expressed a spotless phenotype that was the same as the donor strain, even after they grew more than twice as large as the original size (Kawamura and Watanabe, 1984). This result is consistent with the notion that bud growth does not need the supply of parent (host) epithelial tissues.
Both results of P. stolonifera and P. misakiensis strongly suggest that epithelial components of a new blastogenic generation are all derived from a relatively small number of cells of the bud primordium. In other words, budding involves the removal of a large number of somatic cells and tissue of the parent animal. We would like to emphasize, therefore, that asexuals as well as sexuals have a good chance to purge harmful mutations.
Genetic variation in sexual reproduction
Genetic variation is undoubtedly the motive force of biological evolution. It can be promoted by chromosomal recombination (Barton and Charlesworth, 1998), shuffling of male and female genomes (Maynard Smith, 1978), neutral or beneficial mutations by nucleotide substitution (Kimura, 1967; Johnson, 1999), and genomic change of larger scale such as duplication or deletion (Ohno, 1970; Hughes, 1994; Wolfe and Shields, 1997; Holland et al., 1994). Sexual reproduction has the privilege of executing both the recombination and shuffling of genomes. It has been believed that those recombination and shuffling are beneficial by allowing favorable alleles to come together (Fisher, 1930). At the same time, however, it is also possible that favorable sets of genes having been accumulated through natural selection are broken up by genetic recombination (Barton and Charlesworth, 1998). The compromise is that recombination can be selectively advantageous if different gene combinations are favored in different generations and in different circumstances (Maynard Smith, 1978). In any case, sex offers some efficient methods for genetic variations without harmful mutations. Clonal reproduction either by parthenogenesis or asexual reproduction does not have such convenient methods, which is one of major reasons why clonal reproduction is thought to be disadvantageous to long-term survival (Barton and Charlesworth, 1998).
How fast are genetic variations fixed in the genome? The mutation rates seem to be determined by a balance between natural selection favoring lower mutation rates and opposing selective forces favoring higher mutation rates (Dawson, 1998). They vary widely among different species (Drake, 1991). In Escherichia coli, the rate has been estimated as 1.7×10−4/haploid genome/generation (Kibota and Lynch, 1996). In eukaryotes, it is 0.84 in Daphnia (Deng and Lynch, 1997), 0.3 – 0.4 in Drosophila melanogaster (Mukai et al., 1972; Keightley, 1994), about 0.1 in inbred population of mice (Caballero and Keightley, 1998), and about 5 in human (Kondrashov and Crow, 1993).
Does clonal population give rise to genetic variation ?
Some aphids in Australia exhibit a complete absence of sexual reproduction. These wild-living parthenogenetic lineages have genetic variations in microsatellites and a few other inheritable components (Wilson et al., 1999). A clone of laboratory-maintained parthenogenetic aphids was examined genetically over 32 generations (Lushai et al., 1998). A putative germline mutation was noted once and somatic mutations were noted four times. Mitotic unequal crossing over seems to occur in X chromosome (Mandrioli et al., 1999). A greenbug in the United States reproduces primarily by apomictic parthenogenesis, which is interrupted by a periodic sexual cycle. Shufran et al. (1997) have shown that the intergenic spacer of the rDNA is stable within parthenogenetic clones and that periodic sexual reproduction is a primary mechanism for the generation and maintenance of genetic variability.
In P. misakiensis, two examples of genetic variation have been known so far. One is tunicate C-type lectins of 14 kDa (TC14) (Suzuki et al., 1990). C-type lectins are calcium-dependent carbohydrate-recognition proteins (Drickamer, 1993). They have a common sequence motif of 115 to 130 amino acid (aa) residues. TC14 consisting of 125 aa contains only the carbohydrate recognition domain that binds to D-galactose (Suzuki et al., 1990) and D-fucose (Poget et al., 1999). A cDNA encoding another type of TC14 has been isolated (Shimada et al., 1995), named TC14-2 in relation to the original one that was renamed TC14-1. Two additional isoforms have recently been purified (TC14-3, Matsumoto et al., in preparation; TC14-4, C. Nakamoto, unpublished data). Four TC14s are all translated and have the conserved half cysteine and carbohydrate-binding amino acids (Fig. 4A). We found that although they are redundant, their functions are not the same. TC14-3 showed the highest activity of cell coagulation (Matsumoto et al., in preparation).
Fig. 4
Two examples of polymorphism in Polyandrocarpa genes. (A) Four isoforms of TC 14 lectins from white spot strain. Red arrow shows the N-terminus of mature secretory proteins. Blue letters show carbohydrate-binding amino acids. (B) Four isoforms of P-serpins from white spot strain. Red letters show six, nine or 18 nucleotide inserts.

Polyandrocarpa serine protease inhibitor (P-serpin) is another example of genetic variation (Kawamura et al., 1998). It belongs to a secretory type of polypeptide, referred to as Kazal's inhibitor (Kazal et al., 1948) that is characterized by a peculiar consensus signature with conserved half-cystines (Greene and Bartelt, 1969). Our project of cDNA analysis has shown that P-serpin is expressed enormously during budding (Kawamura et al., 1998). P-serpin contained two related cDNAs, P-serpin 1 discernible from P-serpin 2 by six nucleotide longer than the latter (Fig. 4B). Both P-serpin 1 and 2 had an elastase inhibitor domain and a trypsin inhibitor domain, showing homology to ovomucoid, double-headed pro-tease inhibitor, pancreatic secretory trypsin inhibitor and acrosin inhibitor, all of which belong to Kazal's inhibitor family. Recently, at least two additional subtypes of P-serpin were found (Y. Kariya, unpublished data). They had an insert of either nine or 18 nucleotides long without frame-shift (Fig. 4B). Genetic variants were also found within P-serpin 2. A total of 59 P-serpin 2 cDNAs examined could be classified into four groups, each having the substitution of two or three nucleotides (our unpublished data).
Gene duplication is an important source of evolutionary novelty. Most duplications are of just a single gene, but Ohno (1970) has proposed that whole-genome duplication (polyploidy) is an important evolutionary mechanism. The genetic redundancy of TC14 and P-serpin may be ascribed in part to ancient duplication of their genes or of the entire genome. Many duplicate genes have been found in the yeast Saccharomyces cerevisiae (Kaback, 1995). Wolfe and Shields (1997) have proposed a model in which this species is a degenerate tetraploid resulting from a whole-genome duplication that occurred after the divergence of Saccharomyces from Kluyveromyces. In P. misakiensis, it is possible to assume that the genetic variation may have also come from neutral or less harmful mutations accumulated during asexual generations, as suggested by nucleotide substitutions in P-serpin 2.
Sniegowski (1997) predicts that optimal mutation rates will evolve only in asexual populations. In Polyandrocarpa, as mentioned earlier, buds are endowed with a small number of parental epithelial cells during their growth (cf., Figs. 2,3). If only normal cells are transferred to the bud from the parent that is a carrier of deficient genes, the mutation will be extinguished in the next asexual animal (Fig. 2A). On the other hand, if some of those cells transferred have a deficient gene, the mutation will be emphasized in the subsequent asexual generations, thus fixing genetic polymorphism very promptly in a clonal population. Therefore, our conclusion is that asexual reproduction can give rise to genetic variation.
Is cell renewal the privileged phenomenon for sexual reproduction?
Unlike immortal germ cells, normal somatic cells have a finite life span. Nonetheless, nuclear transplantation experiments have shown that somatic cell nuclei are capable of resetting the developmental clock and resuming ontogeny after introduced into enucleated oocytes of frogs (King and Briggs, 1956; Gurdon, 1962), sheeps (Wilmut et al., 1997) and mice (Wakayama et al., 1998). This reversible aging of somatic cell nuclei has recently been accounted for in relation to the structure of chromosomal extremities (telomeres). Cell division accompanies the progressive shortening of telomeres (Harley et al., 1990; Hastie et al., 1990). It has been first discovered in Tetrahymena that the chromosomal integrity is maintained by telomerase, a ribonucleoprotein that is capable of synthesizing telomeric repeats (Greider and Blackburn, 1985, 1987). Telomerase is a kind of terminal transferase (Morin, 1989) with the activity of reverse transcriptase (Lingner et al., 1997). It is expressed strongly in germline of Xenopus (Mantell and Greider, 1994) and human (Wright et al., 1996), in immortal and cancer cells (Kim et al., 1994) and in stem cells such as hematopoietic progenitors (Chiu et al., 1996). On the other hand, it appears to be stringently repressed in normal human somatic cells (Kim et al., 1994). Telomerase-deficient mouse cells showed chromosomal abnormalities including end-to-end fusion (Blasco et al., 1997). When the catalytic subunit of human telomerase is expressed in telomerase-negative normal cells, the telomerase-expressing clones can extend the length of telomeric DNA and exceed their life-span (Bodnar et al., 1998, Vaziri and Benchimol, 1998). These results provide evidence for the telomere hypothesis that telomere shortening is the molecular clock that triggers replicative senescence (Harley, 1991).
Sexual reproduction appears to reset telomere shortening at each generation, as telomerase is expressed in germline. It is quite in a mystery if telomerase also operates in asexual reproduction, but our recent EST (expressed sequence tag) data suggested that it might be the case. Out of about 2,000 ESTs in P. misakiensis (Kawamura et al., 1998 and our unpublished data), four matched to retrotransposon and retroviral reverse transcriptase with very high similarity (Table 1), although full length of cDNAs remains to be analyzed. As already mentioned, telomerase has the enzyme activity of reverse transcriptase related to retrotransposon and retroviral reverse transcriptase (Cech et al., 1997).
Table 1
ESTs coding for possible nuclear proteins in P. misakiensis.

Another mechanism is known to influence cell aging in asexual reproduction of P. misakiensis. We would like first to refer to a dedifferentiation system for asexual reproduction (Fig. 5A). In botryllid and polystyelid tunicates, a bud consists of outer and inner epithelia (Kawamura and Watanabe, 1982a, b), of which the inner epithelium evaginates or invaginates to form various organs such as the brain, pharynx and gut (Kawamura and Nakauchi, 1984; Kawamura and Sugino, 1999). It is evident, therefore, that the inner, atrial epithelium of the bud is developmentally multipotent (Kawamura and Nakauchi, 1984; Fujiwara and Kawamura, 1992). In P. misakiensis, the inner, multipotent epithelium expresses several differentiation markers such as pigment granules and alkaline phosphatase in the cytoplasm or on the cell surface (Fujiwara and Kawamura, 1992, Kawamura and Fujiwara, 1994). The cell has a prolonged doubling time of more than 170 hrs (Kawamura et al., 1988). At stages of morphogenesis, on the other hand, cells of organ placodes lose the differentiation markers and enter rapid cell cycling of about 13 hrs, indicating that transdifferentiation should occur (Fujiwara and Kawamura, 1992; Kawamura and Fujiwara, 1994).
Fig. 5
Dedifferentiation systems of multipotent cells in P. misakiensis. (A) Comparison of a dedifferentiation system with a stem cell system. (B) Primary structure of TRAMP protein involved in dedifferentiation. 1, signal peptide; 2, 7, Scavenger receptor; 3, 4, 6, Low density lipoprotein receptor; 5, kringle domain-like hairpin loop; 8, serine protease. (C) Circuit of differentiation and dedifferentiation of multipotent cells regulated by TC lectin and TRAMP in culture.

In hydras (Bosch and David, 1991) and planarians (Baguñà et al., 1989; Shibata et al., 1999), stem cell systems are used for asexual reproduction and regeneration (Fig. 5A). They are very sensitive to ionizing radiation (Fradlin et al., 1978; Baguñà et al., 1989). In P. misakiensis, on the other hand, irradiated colonies resume bud formation and bud development in temporal accordance with the restoration of mitotic activity even after irradiation at 80 Gy (Kawamura et al., 1995). This remarkable resistance to ionizing radiation affords another evidence for dedifferentiation system in asexual reproduction of Polyandrocarpa.
Several factors have been identified to regulate dedifferentiation and cell division of multipotent cells in P. misakiensis. One is TC14 lectins. Mature proteins are secreted into the hemolymph during budding and cause mesenchymal cell aggregation to form epithelial tissues (Kawamura et al., 1991). As already mentioned, our published and unpublished studies revealed four kinds of isoforms (TC14-1, TC14-2, TC14-3, TC14-4) (Suzuki et al., 1990; Shimada et al., 1995). They form homo- or heterodimers (Poget et al., 1999). Heterodimers of TC14-2 and TC14-3 isolated from living animals exhibited a remarkable effect on multipotent cells in vitro (Matsumoto et al., in preparation). When cells growing at log phase were treated with TC lectins, they became swollen, ceased to divide and expressed the inner epithelium-specific antigen. This activity of the lectins was blocked by D-galactose. By preparing recombinant proteins, Matsumoto et al. (in preparation) found that only TC14-3 caused cell cycle arrest and induced cell differentiation. These results have shown that TC14-3 acts as autocrine factor to give replicative senescence to the multipotent epithelium. Interestingly, C-type lectins that TC14s belong to have the three-demensional structure related to angiogenesis inhibitor endostatin, although almost no sequence similarity has been found (Hohenester et al., 1998).
A few factors that counteract with TC lectins have been known so far in P. misakiensis. Both retinoic acid (RA) and proteases are able to induce in vivo the secondary bud axis when applied to growing buds (Hara et al., 1992; Kawamura and Watanabe, 1987; Kawamura et al., 1993). Although RA did not directly cause in vitro dedifferentiation of the atrial epithelium, the conditioned medium of RA-treated mesenchymal cells caused dedifferentiation of explants of the atrial epithelium (Kawamura et al., in preparation). The conditioned medium showed the increased activity of trypsin-like serine protease (Kawamura and Fujiwara, 1995). RA is one of endogenous retinoids in P. misakiensis (Kawamura et al., 1993). In developing buds, the enzymatic activity of possible retinoic acid synthase is expressed in the epidermis around the dedifferentiating tissue (Kawamura et al., 1993). Both retinoic acid receptor (Hisata et al., 1998) and retinoid X receptor (Kamimura et al., 2000) are induced to express in mesenchymal cells by RA. These results suggested that RA is the primary signal to trigger bud development. RA-treated mesenchymal cells might secrete the secondary signal to induce dedifferentiation of the atrial epithelium.
Recently, cDNA fragments have been isolated from RA-treated mesenchymal cells by differential display technique (Ohashi et al., 1999). One of them had sequence similarity to trypsin. A full-length cDNA had a serine protease domain at the C-terminus of deduced amino acid sequence (Fig. 5B). At the N-terminus, on the other hand, multiple functional domains were located. The boundary between N- and C-termini had the recognition site cut by plasmin, plasminogen activator and other serine proteases, suggesting that the protein attains to functional maturity after enzymatic modification. Thus, this multi-functional protein is called tunicate retinoic acid-inducible modular protease (TRAMP) (Ohashi et al., 1999).
Recombinant protein of the TRAMP catalytic domain has promoted cell growth and cell motility in a dose-dependent manner (Ohashi et al., 1999). Cells became smaller and lost cytoplasmic granules, suggesting that dedifferentiation might occur. TRAMP showed trypsin-like enzyme activity, while authentic trypsin did not show any mitogenic activity (Ohashi et al., 1999). This result suggested that the sequence responsible for the mitogenic activity should be located outside the conserved region of serine proteases.
Urokinase-type plasminogen activator (uPA) is the well-known vertebrate protease that has the mitogenic activity (Kirchheimer et al., 1988). Human uPA consists of 411 amino acids except for signal peptide. Like TRAMP, it is a modular protease with EGF-like domains and kringle motifs at the N-terminus and with a catalytic domain at the C-terminus (Leslie et al., 1990). Many investigators agree with the notion that the mitogenic activity of uPA is independent of the enzymatic activity (Kirchheimer et al., 1987). Glycosylated EGF and/or kringle domains of uPA at N-terminus have the mitogenic effect on melanoma cells (Koopman et al., 1998) and osteosarcoma cells (Kirchheimer et al., 1987). In these cases, the growth signal of uPA is mediated by high affinity uPA receptors (Rabbani et al., 1992). On the other hand, a N-terminal polypeptide is insufficient for the growth of normal fibroblasts (De Petro et al., 1994), and a full length of uPA is required by the vascular smooth muscle cells (Kanse et al., 1997). Here, uPA seems to use a signal transduction system other than the high affinity receptor. A similar system may work on tunicate cells, as TRAMP recombinant proteins lacking the N-terminal region still have growth-promoting activity.
In the dedifferentiation system of P. misakiensis, the multipotent epithelium is regulated to differentiate by counteracting factors such as TC14 lectins and serine protease (Fig. 5C). In the quiescent state or in the presence of TC14s, multipotent cells have the doubling time of more than 170 hr, at least 13-fold longer than that of dedifferentiated cells (Kawamura et al., 1988). We assume that this slow cell cycling of the multipotent epithelium is practically effective in suppressing cell aging.
Conclusion
Unlike empirical hypotheses, we could not find any serious demerits of asexual reproduction for survival in P. misakiensis. Asexual reproduction involves the removal of a large number of somatic cells in the process of budding. Clonal animals budded have recognizable genetic variations including gene redundancy and polymorphism. A few polypeptides can regulate differentiation states of the multipotent cells, which would serve to retard their aging. It is interesting to ask how many years hereafter the clones of P. misakiensis can continue their asexual lives. We are also interested in the difference in genomic composition between the subclone of Usa and that of Shimoda that have been isolated from each other for just 20 years.
Acknowledgments
We would like to thank Dr. T. Yubisui for encouraging us during the course of study. Thanks are also due to J. Matsumoto and Y. Kariya for the permission of use of a part of their unpublished data. This work was supported in part by Grant-in-Aid to KK (11480221) and SF (11780535) from the Ministry of Education, Sports, Culture and Science of Japan.