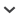Epidermal growth factor receptor (EGFR) is overexpressed in human pancreatic cancer and is one of the clinical targets in its treatment. In the present study we investigated the mechanism underlying ultraviolet C (UVC)-radiation-induced cell growth inhibition and downregulation of EGFR in human pancreatic cancer cells (Panc1 and KP3). The cell proliferation assay indicated that Panc1 and KP3 cells were more sensitive to UVC radiation, and their growth was significantly inhibited compared to cells of the normal human pancreatic epithelial cell line, PE. Although EGFR levels was extremely low in PE cells, EGFR were highly overexpressed in Panc1 and KP3 cells, and UVC radiation downregulated the expression of EGFR in a time-dependent manner and induced phosphorylation of EGFR at Ser1046/1047 (S1046/7) in Panc1 and KP3 cells. UVC radiation induced activation of p38 mitogen-activated protein kinase (MAPK), and EGFR phosphorylation at S1046/7 induced by UVC radiation was markedly attenuated by the inhibition of p38 MAPK. Moreover, fluorescence microscopy revealed that p38 MAPK activated by UVC radiation triggered EGFR internalization and that this was not correlated with c-Cbl, an ubiquitin ligase, which plays an important role in EGF-induced EGFR downregulation. Taken together, our results suggest that in pancreatic cancer cells UVC radiation induced desensitization of the cells to EGFR stimuli via phosphorylation of EGFR at S1046/7 by activation of p38 MAPK, independent of c-Cbl.
INTRODUCTION
Pancreatic cancer is a highly lethal disease that is usually diagnosed at a late stage. Gemcitabine is now regarded as the first-line agent for treatment of advanced pancreatic cancer, but the median survival time of patients treated with gemcitabine is not satisfactory. Therefore, researchers expanded their interest to the development of new treatments for inoperable pancreatic cancer. Epidermal growth factor (EGF) receptor (EGFR) has been shown to be overexpressed in pancreatic cancer 1–3. In general, binding of specific ligands such as EGF and transforming growth factor (TGF)-α to the extracellular domain results in EGFR dimerization and autophosphorylation of the tyrosine kinase domain, leading to the activation of downstream signaling pathways that are involved in cell proliferation and survival 4. Recently, the addition of the EGFR-targeted therapy to gemcitabine in the treatment of advanced pancreatic cancer has been demonstrated to provide a significant survival benefit 5.
With regard to desensitization, receptor downregulation is the most prominent regulatory system of EGFR signal attenuation and involves the internalization and subsequent degradation of the activated receptor in the lysosomes 6. Based on the current knowledge of the mechanism underlying EGFR desensitization, this molecular event seems to involve several important phosphorylation sites. One is the phosphorylation at Tyr1045, which provides a docking site for the ubiquitin ligase c-Cbl, resulting in ubiquitination of the EGFR 7, and the others are the phosphorylation at serine or threonine residues, which are thought to represent a mechanism for attenuation of the receptor kinase activity 8–10. Among the major sites of serine and threonine phosphorylation of the EGFR, it has previously been shown that the serine 1046/1047 (S1046/7) phosphorylation sites are required for EGFR desensitization in EGF-treated cells 10. In addition, we have recently reported that p38 mitogen-activated protein kinase (MAPK) controls EGFR desensitization via phosphorylation at S1046/7 11, suggesting that serine phosphorylation of EGFR or p38 MAPK activation might be considered to be a new therapeutic target, especially to counter cancer cells in the colon, lung, breast and pancreas that highly express EGFR.
Ultraviolet (UV) radiation from sunlight is classified into three different wavelength ranges: long-wavelength UVA (320–400 nm), medium-wavelength UVB (280–320 nm) and short-wavelength UVC (200–280 nm). Whereas UVA radiation does not directly excite DNA, UVB and UVC radiation do and cause mostly pyrimidine dimers 12. However, UVC radiation does not actually reach the Earth's surface since it is filtered out by the atmosphere. Therefore, UVA and UVB radiation are recognized as the major carcinogenic components of sunlight 13. The possibility of the application of UVC radiation for treatment of human cancer has been demonstrated 14, 15, but its precise mechanism has not been fully elucidated. It has previously been reported that UV radiation causes EGFR arrest in early endosomes and that its internalization is instigated by p38 MAPK 16. In that study, the underlying mechanism entailed phosphorylation of EGFR at a short segment (amino acids 1002–1022) containing multiple serines and threonines. It has also been reported that UV light reduces the proliferative potential of skin-derived cancer cell lines, thus suggesting that it would be particularly beneficial to inhibit the growth of EGFR-overexpressing cells 14. In the present study, we used UVC radiation as a tool to activate p38 MAPK and investigated the effect of UVC radiation on cell proliferation of human pancreatic cancer cells. We demonstrated here that UVC radiation inhibited cell growth with downregulation of EGFR through its phosphorylation at S1046/7 by activation of p38 MAPK. This UVC-radiation-induced downregulation of EGFR was independent of c-Cbl, which is required for EGF-induced EGFR downregulation.
MATERIALS AND METHODS
Materials
The p38 MAPK-selective inhibitors SB203580 and BIRB0796 were obtained from Calbiochem-Novabiochem Co. (La Jolla, CA) and Dr. Philip Cohen (University of Dundee, UK), respectively. Concanavalin A (Con A) was obtained from Sigma Chemical Co. (St. Louis, MO). Antibodies against total EGFR and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against phospho-EGFR (Tyr 992, Tyr1045, Tyr1068, Tyr1173 and S1046/7), phospho-p44/p42 MAPK, p44/p42 MAPK, phospho-p38 MAPK, p38 MAPK, phospho-stress-activated protein kinase/c-Jun-N-terminal kinase (SAPK/JNK), SAPK/JNK, phospho-Akt, Akt, EEA-1 and c-Cbl were purchased from Cell Signaling (Beverly, MA). The ECL Western blot detection system was purchased from GE Healthcare (Buckinghamshire, UK). The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT) cell proliferation kit I and Cell Proliferation ELISA (BrdU) were obtained from Roche Diagnostics Co. (Indianapolis, IN). Alexa Fluor 546® conjugated donkey anti-mouse IgG antibodies and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Invitrogen and Wako (Tokyo, Japan), respectively. Predesigned siRNAs targeting p38 MAPK (On-TARGET plus Duplex J-003512-20, Human MAPK14) was purchased from Thermo Fisher Scientific K.K. (Yokohama, Japan). Sequences are as follows: sense: GGAAUUCAAUGAUGUG UAUUU and antisense: AUACACAUCAUUGAAUUCCUU. Other materials and chemicals were obtained from commercial sources.
Cell Culture
Panc1 and KP3 pancreatic cancer cells were provided from American Type Culture Collection (ATCC; Manassas, VA). Primary normal human pancreatic epithelial (PE) cells were maintained in Cell Systems Corporation (CS-C) medium purchased from DC Pharma Biomedical (Kirkland, WA). Panc1 and KP3 cells were grown in RPMI 1640 medium (Invitrogen, San Diego, CA) supplemented with 10% heat-inactivated fetal calf serum (FCS), penicillin (100 U/ml) and streptomycin (100 µg/ml) (all from Invitrogen) in a humidified 95% air/5% CO2 incubator at 37°C.
UVC-Radiation Exposure
Exposure of cells to UVC radiation was performed in a UVC 500 UV Crosslinker (GE Healthcare) with 0–500 J/m2 (J) UV radiation at 254 nm. After aspiration of the growth medium, the cells were exposed to UVC radiation (0, 100, 200, 300, 400 and 500 J) and then incubated in growth medium for 0, 1, 5, 10, 20, 30, 60 and 120 min or 12, 24 and 48 h.
Cell Viability Assay
In the cell viability assay, cells (5 × 103/well) were seeded onto 96-well plates and 24 h later were exposed to 0, 100, 200, 300, 400 or 500 J of UVC radiation. The cells were then incubated in RPMI 1640 or CS-C medium in a humidified 95% air/5% CO2 incubator at 37°C for 48 h. The remaining cells were counted using an MTT cell proliferation kit in accordance with instructions of the manufacturer. All assays were done in triplicate.
BrdU Incorporation Assay
Cells (7 × 103/well) were seeded onto 96-well plates and 24 h later were exposed to 0, 100, 200, 300 or 400 J of UVC radiation. The cells were then incubated in RPMI 1640 or CS-C medium with 1% FCS for 24 h. BrdU incorporation was measured using a Cell Proliferation ELISA. All assays were done in triplicate.
Western Blot Analysis
The cells were lysed in lysis buffer [20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 50 mM NaF, 50 mM Hepes, 1 mM Na3VO4 and 2 mM phenylmethylsulfonyl fluoride (PMSF)] and scraped from the petri dishes. Protein extracts were examined by Western blot analysis as described previously 17. The protein was electrophoresed and transferred onto an Immune-Blot PVDF Membrane (Bio-Rad, Hercules, CA). Membranes were blocked with 5% fat-free dry milk in phosphate-buffered saline (PBS) containing 0.1% Tween-20 (PBS-T) for 30 min before 16 h incubation with primary antibodies (1∶1000 dilution). Peroxidase-labeled antibodies raised in goat against rabbit IgG were used as secondary antibodies (1∶2000 dilution; 1 h incubation). Peroxidase activity on the membrane was visualized on X-ray film by means of the ECL Western blot detection system.
Immunofluorescence Microscopy
Immunofluorescence microscopy was performed as described previously 18. The cells grown on cover slip-bottom dishes were first treated with or without SB203580 (20 µM) or Con A (100 µg/ml) for 1 h at 37°C, followed by exposure to anti-EGFR antibodies for 15 min in RPMI 1640 medium containing 1% bovine serum albumin (BSA). They were then exposed to UVC radiation (200 J) and incubated in RPMI 1640 medium with or without SB203580 for an additional 30 min. They were then fixed with 4% paraformaldehyde for 10 min on ice and then exposed to 0.1% Triton X-100 for 10 min to permeabilize the cell membrane. They were then exposed to anti-phospho-EGFR (S1046/7), anti-c-Cbl or anti-early endosome antigen (EEA)-1 antibodies, followed by exposure to Alexa Fluor 488® conjugated goat anti-rabbit IgG, Alexa Fluor 546® conjugated anti-mouse IgG antibodies and DAPI for 1 h. The cells were then examined by fluorescence microscopy using a Biorevo BZ-9000 (Keyence, Tokyo, Japan) according to the manufacturer's protocol.
siRNA Protocol
Transfection was performed according to the manufacturer's protocol (Bio-Rad, Tokyo, Japan). In brief, 5 µl of siLentFect (Bio-Rad) and finally 100 nM of siRNA were diluted with Opti-MEM, preincubated at room temperature for 20 min, and then added to the Opti-MEM without FCS. Cells were incubated at 37°C for 24 h with siRNA–siLentFect complexes. The medium was then changed to fresh RPMI 1640 medium containing 10% FCS, and cells were incubated for additional 24 h and subsequently harvested for Western blot analysis.
Quantification of Cell Surface EGFR by Enzyme-Linked Immunosorbent Assay (ELISA)
Quantification of cell surface EGFR was performed as described previously 18. In brief, Panc1 and KP3 cells were pretreated with the indicated compounds and then exposed to the mouse anti-EGFR antibody (Santa Cruz Biotechnology) that recognizes the extracellular domain of the EGFR (1∶50 dilution) in DMEM containing 1% bovine serum albumin (BSA) for 15 min at 37°C. The cells were then incubated for the indicated times after exposure to UVC radiation (200 J) and then fixed with 4% paraformaldehyde for 10 min on ice. After blocking with 1% BSA in PBS for 1 h, the cells were exposed to an anti-mouse IgG, horseradish peroxidase-linked whole antibody (GE Healthcare, Piscataway, NJ) for 1 h at room temperature, followed by washing four times with PBS containing 1% BSA. Finally, the cells were exposed to 50 µl of 1-Step Ultra TMB-ELISA reagent (Pierce, Rockford, IL) for 5 min at room temperature. Then 50 µl of 2 M sulfuric acid was added to each well to stop the reaction. The absorbance of each sample at 450 nm was then measured.
Densitometric Analysis
The densitometric analysis was performed using scanner and image analysis software (ImageJ ver. 1.32). The background-subtracted signal intensity of each protein signal was normalized to the respective control signal. All data were obtained from at least three independent experiments.
RESULTS
Effect of UVC Radiation on Cell Proliferation and Downregulation of EGFR in Human Pancreatic Cancer Cells Compared to PE Normal Human Pancreatic Epithelial Cells
We first performed a cell viability assay to examine the suppressive effect of UVC radiation in Panc1 and KP3 pancreatic cancer cells compared to PE normal human pancreatic epithelial cells. The IC50 value of UVC radiation in KP3 cells was approximately 150 J, whereas that in Panc1 cells was approximately 200 J (Fig. 1A). In contrast, the IC50 value of UVC radiation in PE cells was over 500 J (Fig. 1A), and statistical analysis revealed significant differences between the pancreatic cancer cells and PE cells (see figure legends).
FIG. 1.
Panel A: Inhibition of viability of Panc1, KP3 and PE cells by UVC radiation. A cell viability assay was performed using the MTT cell proliferation kit I. Incubation time after UVC irradiation was 48 h in the MTT assay. Results are expressed as the percentage of growth, with 100% representing untreated control cells. ▴, Panc1 pancreatic cancer cells; •, KP3 pancreatic cancer cells; □, PE normal pancreatic epithelial cells. Panel B: Inhibition of cell proliferation by UVC radiation in Panc1, KP3 and PE cells. The measurement of BrdU incorporation during DNA synthesis was performed using cell proliferation ELISA (BrdU). Incubation time after UVC irradiation was 24 h in the BrdU assay. Results are expressed as the percentage of growth, with 100% representing untreated control cells. Bars are SD of triplicate assays. The asterisks (*, **) indicate statistically significant increases (P < 0.05) compared to the corresponding viability of PE cells.

We next investigated the effect of UVC radiation on cell proliferation using BrdU, an analog of thymidine. In Panc1 and KP3 cells, UVC radiation caused a marked inhibition of BrdU incorporation (Fig. 1B). The inhibitory effect in these cells appeared clearly at 100 J (40% reduction compared to the control), and 200 J of UVC radiation caused an approximately 65% reduction in BrdU incorporation (Fig. 1B). In PE cells, 100 J of UVC radiation did not influence BrdU incorporation, and even 200 J of UVC radiation caused less reduction (about 25%) than seen in pancreatic cancer cells. Taken together, these results suggest that UVC radiation has a potent anti-proliferative effect on Panc1 and KP3 pancreatic cancer cells, although PE normal pancreatic epithelial cells were more resistant to UVC radiation.
It has previously been reported that overexpression of EGFR in pancreatic cancers has an important role in its aberrant growth and progression 19. Therefore, we examined the expression level of EGFR protein in these cells. We found that EGFR was highly expressed in Panc1 and KP3 cells, whereas very low levels of EGFR were observed in PE cells (Fig. 2A). We next examined the effect of UVC radiation on the protein level of EGFR in pancreatic cancer cells and found that exposure to UVC radiation significantly decreased the level of EGFR in a time-dependent manner in Panc1 and KP3 cells (Fig. 2B and C). Although it has previously been reported that UV radiation induces endosome arrest of EGFR in HeLa epidermal carcinoma cells 20, we found that UVC radiation induced downregulation of EGFR in pancreatic cancer cells. EGFR protein levels in PE cells were not affected by UVC radiation (Fig. 2B and C). These results also led us to speculate that UVC-radiation-induced inhibition of cell growth might be due at least in part to the decrease in EGFR protein levels in Panc1 and KP3 pancreatic cancer cells.
FIG. 2.
Panel A: Expression level of EGFR protein in Panc1, KP3 and PE cells. The cells were harvested and protein extracts (5 µg each) were analyzed by Western blotting. The upper and middle panels present the results after short and long exposure to film, respectively. Panel B: UVC radiation caused a decrease in EGFR protein levels in Panc1, KP3 and PE cells. The cells were exposed to UVC radiation at 200 J and then incubated for the indicated periods. Protein extracts were then harvested and examined by Western blotting using anti-EGFR and anti-GAPDH antibodies. Panel C: Quantification of the Western blotting results shown in Fig. panel A. The asterisks (*, **) indicate statistically significant difference (P < 0.05) compared to PE cells.

Effect of UVC Radiation on the Phosphorylation of EGFR at S1046/7 in Human Pancreatic Cancer Cells
Since we observed that exposure to UVC radiation decreased the protein level of EGFR (Fig. 2B and C) in Panc1 and KP3 cells, we next investigated its effect on the phosphorylation of EGFR. UVC radiation caused significant increase in the phosphorylation level of EGFR at S1046/7 that was observed 10 min after exposure and reached a maximum at 60 min (Fig. 3A and B). However, UVC radiation had little effect on EGFR phosphorylation at Tyr 992, 1045, 1068 or 1173 (Fig. 3A and B), all of which are well known to be autophosphorylation sites 7, 21–23. This observation was consistent with the previous study 20. Thus it is likely that EGFR downregulation does not require tyrosine phosphorylation and that phosphorylation of EGFR at S1046/7 might play a role in UVC-radiation-induced EGFR downregulation in pancreatic cancer cells.
FIG. 3.
UVC radiation caused the phosphorylation of EGFR at S1046/7 in pancreatic cancer cells. Panc1 (panel A) and KP3 (panel B) cells were exposed to UVC radiation at 200 J and then incubated for the indicated periods. Protein extracts were then harvested and examined by Western blotting using anti-phospho-EGFR at S1046/7, Y992, Y1045, Y1068 and Y1173, anti-EGFR and anti-GAPDH antibodies. The each lower bar graph shows quantification data for the phosphorylation levels of EGFR after normalization with respect to GAPDH.

Effects of UVC Radiation on the MAPK and Akt Pathways in Human Pancreatic Cancer Cells
To elucidate how UVC radiation causes EGFR phosphorylation at serine residues, we next examined the effects of UVC radiation on the activation of kinase cascades. We found that UVC radiation induced activation of p44/p42 MAPK, SAPK/JNK, p38 MAPK and Akt in Panc1 cells (Fig. 4). Similar effects were also seen in KP3 and PE cells (data not shown). These results led us to further investigate which kinase plays a critical role in the phosphorylation of EGFR at serine residues induced by UVC radiation in pancreatic cancer cells.
FIG. 4.
UVC radiation caused the phosphorylation of p44/p42 MAPK, p38 MAPK, SAPK/JNK and Akt in pancreatic cancer cells. Panc1 cells were exposed to UVC radiation at 200 J and then incubated for the indicated periods. Protein extracts were then harvested and examined by Western blotting using anti-phospho-p44/p42 MAPK, p44/p42 MAPK, phospho-p38 MAPK, p38 MAPK, phospho-SAPK/JNK, SAPK/JNK, phospho-Akt, Akt and GAPDH antibodies.

Effect of p38 MAPK on UVC-Radiation-Induced Phosphorylation of EGFR at S1046/7 in Human Pancreatic Cancer Cells
We used several inhibitors to investigate the upstream signaling of the phosphorylation of EGFR at S1046/7 by UVC radiation in Panc1 and KP3 cells. As shown in Fig. 5A and B, we found that SB203580 and BIRB0790, both of which are p38 MAPK-selective inhibitors 24, 25, significantly suppressed the phosphorylation of EGFR at S1046/7 induced by UVC radiation in Panc1 and KP3 cells, although it was not suppressed when the cells were pretreated with PD98059, a specific inhibitor of MEK1/2 26, SP600125, a specific inhibitor of SAPK/JNK 27, and Akt inhibitor 28 (data not shown). These results strongly suggest that p38 MAPK is involved in the phosphorylation of EGFR at serine residues induced by UVC radiation. To verify these results, we used gene silencing using p38 MAPK-siRNA on the phosphorylation of EGFR at S1046/7 in these cells. As expected, UVC-radiation-induced phosphorylation of EGFR at S1046/7 was also suppressed by siRNA of p38 MAPK in Panc1 and KP3 cells (Fig. 5C and D), suggesting that phosphorylation of EGFR at S1046/7 by UVC radiation is mediated through the p38 MAPK pathway in pancreatic cancer cells.
FIG. 5.
The inhibition of p38 MAPK suppressed UVC-radiation-induced phosphorylation of EGFR at S1046/7. Panc1 (panel A) and KP3 (panel B) cells were pretreated with 20 µM of SB203580 or 1 µM of BIRB0790 for 1 h, exposed to UVC radiation at 200 J, and then incubated for 1 h. Protein extracts were then harvested and examined by Western blotting using anti-phospho-EGFR at S1046/7, anti-EGFR and anti-GAPDH antibodies. The lower bar graph shows quantification data for the relative phosphorylation levels of EGFR at S1046/7 after normalization with respect to GAPDH. Panels C and D: Effect of gene silencing with p38 MAPK-siRNA transfection on pancreatic cancer cells. Panc1 (panel C) and KP3 (panel D) cells were incubated with 100 nM of p38 MAPK-siRNA or negative control (NC)-siRNA, followed by incubation for 1 h after exposure to UVC radiation (200 J). Protein extracts were then prepared and examined by Western blotting using anti-phospho-EGFR at S1046/7, EGFR, p38 MAPK, p44/p42 MAPK, SAPK/JNK and Akt. An antibody to GAPDH was used as the control for protein loading. The lower bar graph shows quantification data for the relative phosphorylation levels of EGFR at S1046/7 after normalization with respect to GAPDH. Representative results from triplicate independent experiments with similar results are shown. NC indicates negative control.

Effect of the p38 MAPK on the Internalization of EGFR Induced by UVC Radiation in Human Pancreatic Cancer Cells
It is well known that EGF induces internalization of the EGFR via endocytosis and that this is associated with subsequent ubiquitin-mediated degradation of the EGFR 6. Therefore, we next examined whether UVC radiation also induces changes in the cellular localization of the EGFR in pancreatic cancer cells using fluorescence microscopy. When cells were not treated with UVC radiation, fluorescence remained mainly on the cell surface (Fig. 6A, panel 1), and treatment of the cells with UVC radiation (200 J) resulted in the redistribution of the antibody-tagged EGFR to vesicles beneath the plasma membrane 30 min later (Fig. 6A, panel 9). Concurrently, the internalized EGFR induced by UVC radiation was stained with anti-phospho-EGFR (S1046/7) antibodies (Fig 6A, panel 10). Moreover, we demonstrated that this change was virtually blocked when the cells were pretreated with SB203580, a p38 MAPK-selective inhibitor (Fig. 6A, panel 13). We obtained similar results in KP3 pancreatic cancer cells (Fig. 6B). Taken together, our findings strongly suggest that activation of p38 MAPK triggers the change in the cellular localization of EGFR in UVC-irradiated pancreatic cancer cells.
FIG. 6.
EGFR internalization and phosphorylation at S1046/7 induced by UVC radiation were canceled by the treatment with SB203580 in Panc1 (panel A) and KP3 (panel B) pancreatic cancer cells. The cells were pretreated with or without 20 µM SB203580 for 1 h and then labeled with anti-EGFR antibodies. Thirty minutes after exposure to UVC radiation (200 J), they were fixed, permeabilized and treated with anti-phospho-EGFR at S1046/7 antibodies followed by Alexa 546-conjugated secondary antibodies for EGFR (red signal) and Alexa 488 conjugated secondary antibodies for phospho-EGFR at S1046/7 (green signal) and DAPI (blue signal) and then examined by fluorescence microscopy.

To clarify whether or effect of UVC radiation on EGFR is due to internalization, we next measured the amount of EGFR that remained on the cell surface using ELISA. UVC radiation caused a rapid decrease in the amount of cell surface EGFR in a time-dependent manner (Fig. 7A). Moreover, fluorescence microscopy revealed that Con A, an endocytosis inhibitor, inhibited the changes in the localization of the EGFR induced by UVC radiation (Fig. 7B). Taken together, these results strongly suggest that UVC radiation induced EGFR internalization in pancreatic cancer cells.
FIG. 7.
Panel A: UVC radiation induced a decrease in the cell surface amount of EGFR. Line graph shows quantification data for cell surface amounts of EGFR analyzed by ELISA (see the Materials and Methods). ○, Panc1 without UVC irradiation; Δ, KP3 without UVC irradiation; •, Panc1 cells exposed to 30 J of UVC radiation; ▴, KP3 cells exposed to 30 J of UVC radiation. Panel B: EGFR internalization induced by UVC radiation was blocked by treatment with concanavalin A (Con A), an endocytosis inhibitor, in Panc1 cells. The Panc1 cells were pretreated with or without 100 µg/ml of Con A for 1 h and then labeled with anti-EGFR antibodies. After exposure to UVC radiation for 30 min, they were fixed and permeabilized, treated with Alexa 488-conjugated secondary antibodies for EGFR (green signal) and DAPI (blue signal), and then examined by fluorescence microscopy. The asterisks (*, **) indicate statistically significant differences (P < 0.05) compared to PE cells.

Correlation of the Decrease in EGFR Protein Level Induced by UVC Radiation and c-Cbl Binding in Human Pancreatic Cancer Cells
It is well known that c-Cbl plays a role in downregulation of EGFR 6, 29, 30. c-Cbl has been shown to ubiquitinate and downregulate EGFR 7 and was formally established as a RING finger-type E3 ubiquitin ligase 31, 32. Therefore, we next examined the correlation between EGFR and c-Cbl in the UVC-radiation-induced EGFR downregulation. We performed colocalization assays of these internalized EGFRs (Fig. 8A, panels 1, 5 and 9) and c-Cbl (Fig. 8A, panels 2, 6 and 10). Internalized EGFR induced by UVC radiation did not colocalize with the c-Cbl protein (Fig. 8A, panels 9–12), whereas the internalized EGFR induced by EGF was clearly colocalized with c-Cbl (Fig. 8A, panels 5–8), which is consistent with our previous studies 11, 33. Since UVC radiation did not induce the phosphorylation of EGFR at any tyrosine residues, including Tyr1045 (data not shown), it seems unlikely that the internalization of EGFR induced by UVC radiation is associated with c-Cbl. We also performed the colocalization study with these internalized EGFRs and an early endosome marker, EEA-1, to investigate whether there are some differences in the endocytic pathways and found that internalized EGFR induced by either EGF or UVC radiation clearly colocalized with EEA1 (Fig. 8B), suggesting that the mechanisms of endocytosis, at least at the stage of early endosomes, are similar.
FIG. 8.
Panel A: The internalized EGFR induced by UVC radiation is not associated with c-Cbl. Panc1 cells were first labeled with an anti-EGFR antibodies as described above and then treated with EGF (30 ng/ml) or UVC radiation (200 J), followed by fixation and permeabilization. The cells were then exposed to anti-c-Cbl antibodies followed by Alexa 546-conjugated secondary antibodies for EGFR (red signal), Alexa 488-conjugated secondary antibodies for c-Cbl (green signal), and DAPI (blue signal) and then examined by fluorescence microscopy. Panel B: The internalized EGFR was colocalized with EEA-1. Panc1 cells were first labeled with anti-EGFR antibodies then treated with EGF (30 ng/ml) or UVC radiation (200 J), followed by fixation and permeabilization. The cells were then exposed to anti-EEA-1 antibodies, followed by Alexa 546 conjugated secondary antibodies for EGFR (red signal), Alexa 488-conjugated secondary antibodies for EEA-1 (green signal), and DAPI (blue signal) and then examined by fluorescence microscopy. Representative results from at least three independent experiments are shown.

DISCUSSION
Our present study provides first evidence that UVC radiation has a potent anti-proliferative effect, in parallel with inducing EGFR internalization and the subsequent decrease in EGFR protein levels in Panc1 and KP3 human pancreatic cancer cells, whereas little effect on EGFR was observed in PE normal pancreatic epithelial cells (Figs. 1 and 2). However, further investigation is required to prove the correlation of the anti-proliferative effect of UVC radiation and the downregulation of EGFR.
We also found that UVC radiation caused phosphorylation of EGFR at S1046/7, but not tyrosine residues that are well recognized to be autophosphorylation sites of EGFR 7, 21–23, in Panc1 and KP3 cells. Moreover, the internalized EGFR induced by UVC radiation did not colocalize with c-Cbl (Fig. 8A), which reportedly plays an important role in the ubiquitin-mediated degradation induced by EGF 7. These results strongly support our conclusion that the decrease of EGFR protein levels induced by UVC radiation is independent of tyrosine phosphorylation of EGFR in pancreatic cancer cells. We emphasize that UVC radiation can cause EGFR downregulation without tyrosine phosphorylation, which results in cancer cell proliferation 22, 23.
It has previously been demonstrated that EGFR internalization caused by UV radiation is involved in its phosphorylation at a short segment (amino acids 1002–1022) 16. Moreover, mutational removal of serine residues, including 1046 and 1047, was sufficient to abolish UV-radiation-induced internalization 34. In this study, we provide the direct evidence that UVC radiation induces EGFR phosphorylation at S1046/7 via p38 MAPK in pancreatic cancer cells. We also demonstrated that p38 MAPK activated by UVC radiation triggers both of EGFR internalization (Fig. 6) and phosphorylation at S1046/7 (Fig. 5) in Panc1 and KP3 pancreatic cancer cells.
When EGF binds to EGFR molecules on the cell surface, the receptor undergoes dimerization and autophosphorylation at tyrosine residues, and this triggers EGFR-related downstream signaling toward the direction of cell growth 22, 23. On the other hand, the EGFR is also ubiquitinated and internalized into early endosomes, which become late endosomes by a negative feedback system, and eventually the receptors are desensitized 6. We showed here that UVC radiation has little effect on tyrosine phosphorylation (activation) of EGFR. Faint phosphorylation at tyrosine residues is consistent with the decreased BrdU incorporation and the decreased cell number. By contrast, UVC radiation caused phosphorylation of p38 MAPK, resulting in the phosphorylation of EGFR at S1046/7, and subsequently EGFR molecules were internalized into early endosomes and eventually desensitized. A schematic diagram of a hypothetical mechanism by which UVC radiation causes downregulation of EGFR is shown in Fig. 9.
FIG. 9.
A schematic diagram of a hypothetical mechanism by which UVC radiation causes downregulation of EGFR. After EGF binds to EGFR molecules on the cell surface, the receptor undergoes dimerization and autophosphorylation at tyrosine residues, and this triggers EGFR-related downstream signaling 7, 21–23. The EGFR is also ubiquitinated via c-Cbl and internalized into early endosomes, which become late endosomes, and eventually the receptors are desensitized 6, 7. When the cells are treated with UVC radiation, EGFR is not phosphorylated at tyrosine residues, which indicates that UVC radiation fails to induce cell growth signals. In contrast, UVC radiation causes phosphorylation of p38 MAPK, resulting in the phosphorylation of EGFR at S1046/7, and subsequently EGFR molecules are internalized early endosomes. Although the internalized EGFR induced by UVC radiation is not associated with c-Cbl, they are eventually desensitized.

We have previously reported that HSP90 inhibitors as well as (-)-epigallocatechin gallate downregulates EGFR via phosphorylation at S1046/7 by p38 MAPK in human colon and pancreatic cancer cells 33, 35. In addition, there is accumulating evidence that activation of p38 MAPK has a suppressive effect on tumorgenesis 36 and that agents such as gemcitabine 37 and cisplatin 38 can also induce activation of p38 MAPK and internalization of EGFR into endosomal vesicles. Moreover, it has been reported that the S1046/7 phosphorylation sites act to suppress signal transduction for cell growth by the wild-type EGFR 8, 39. While we used UVC radiation as a tool to activate p38 MAPK, we expect to provide a new perspective on cancer therapy by understanding how EGFR phosphorylation at serine residues triggers its downregulation. In addition, the development of devices that supply UVC radiation efficiently is also required for future clinical application. For example, delivery of UVC radiation to the pancreas (e.g. through endoscopic retrograde assisted cholangiopancreatography) could be made intraluminally where dysplasia/small neoplasias at the duct-acinar junctions are detected.
In summary, our results strongly suggest that UVC illumination targets S1046/7 of EGFR that causes the inhibition of pancreatic cancer cell proliferation.
ACKNOWLEDGMENTS
We are very grateful to Ms. Yoko Kawamura for her skillful technical assistance. This work was supported in part by Grant-in-Aid for Scientific Research (22790639 to SA) from the Ministry of Education, Science, Sports and Culture of Japan and a grant by Yokoyama Foundation for Clinical Pharmacology.